Abstract 19082: Cardiac Events and Mortality During Admissions for Delivery in Women with Congenital Heart Disease
Robert M Hayward, Elyse Foster, Zian H Tseng
Circulation. 2014;130:A19082
Abstract
Background: Labor, delivery, and the postpartum period are a time of increased arrhythmia and congestive heart failure (CHF) incidence. With improvements in the treatment of congenital heart disease (CHD), more women are reaching childbearing age and may be at increased risk for cardiac events and mortality during pregnancy and delivery.
Methods: The Healthcare Cost and Utilization Project was used to identify admissions for vaginal and cesarean delivery in California hospitals between 1/1/2005 and 12/31/2011. We compared length of stay, in-hospital mortality, incident CHF, cardiac arrest, and incident arrhythmias for women without CHD to women with non-complex CHD (NC-CHD) and complex CHD (C-CHD).
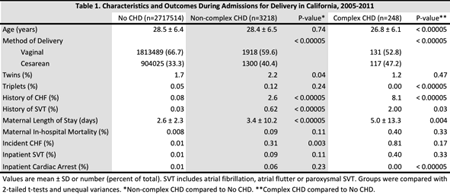
Results: We identified 2,720,980 deliveries resulting in 2,770,382 live births (74% of live births in the state over this period), which included 3,218 women with NC-CHD and 248 women with C-CHD. History of CHF was more common in women with CHD (8.1% for C-CHD, 2.6% for NC-CHD, and 0.08% for women without CHD, p<0.00005 for NC-CHD compared to no CHD and for C-CHD compared to no CHD). Those with CHD were more likely to undergo cesarean section (Table 1). Length of stay was significantly longer in women with CHD (2.6 ± 2.3 days for women without CHD, 3.4 ± 10.2 days for women with NC-CHD and 5.0 ± 13.3 days for women with C-CHD). In-hospital mortality was not significantly higher in women with CHD (Table 1). Incident heart failure, arrhythmias, and cardiac arrest were uncommon in all groups (Table 1).
Conclusions: In this study of 2.7 million women admitted to California hospitals for delivery, women with CHD were more likely to undergo cesarean section and had longer length of stay. Despite more frequent history of CHF in women with CHD, incident CHF and arrhythmias were rare during hospitalization. In-hospital mortality and cardiac arrest were not higher in CHD patients. These results suggest that in pregnant women with CHD, cardiac events and mortality at the time delivery are uncommon.
Article Information
vol. 130 no. Suppl 2 A19082
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Medicine, Div of Cardiology, Section of Cardiac Electrophysiology, Univ of California, San Francisco, San Francisco, CA
- 2Medicine, Div of Cardiology, Univ of California, San Francisco, San Francisco, CA
Abstract 19274: Coronary Artery Ectasia Are Frequently Observed in Patients With Bicuspid Aortic Valves With and Without Aneurysm of the Ascending Aorta
Christine Meindl, Birgit Achatz, Deborah Huber, Ute Hubauer, Stefan Buchner, Kurt Debl, Sabine Fenk, Christina Strack, Christian Hengstenberg, Heribert Schunkert, Christa Meisinger, Lars Maier, Andrea Baessler, Marcus Fischer
Circulation. 2014;130:A19274
Abstract
Background: The exact etiology and prognostic significance of coronary artery ectasia (CAE) is still unknown. The presence of CAE is influenced by genetic factors and related to the presence of aneurysms in other vascular beds. Bicuspid aortic valve (BAV) disease is frequently accompanied by ascending aortic aneurysm. Since the aortic valve, the ascending aorta, and the proximal parts of the coronary arteries share a common embryonic origin, we hypothesized that CAE is associated with BAV disease.
Methods: 181 patients with suspected aortic valve disease (n=101 BAV) underwent both cardiac magnetic resonance (CMR) imaging and coronary angiography. Eighty subjects with tricuspid aortic valve (TAV) disease were similarly studied and served as controls. The readers of the angiograms were blinded to valve type and clinical data. In order to confirm the association of CAE with BAV, the frequency of CAE was evaluated in an in-house BAV registry (n=500) and compared to the frequency of CAE in the German MI family study, in which the heritability of CAE was formerly established (899 with available coronary angiograms), as well as in an observational registry of “real-life patients” undergoing coronary angiography for clinically indicated reasons (n=3471). Furthermore the frequency of CAE was investigated in a subgroup of the KORA MI study, which is a population-based registry that comprises all hospitalized cases of acute non-fatal MI and coronary deaths occurring in inhabitants of a defined study region (n=403).
Results: Compared to TAV disease, CAE occured twice as frequently in CMR confirmed BAV disease, (17.5% vs. 41.6%, p=0.0005). Ascending aortic aneurysm or ectasia was diagnosed in 60 subjects with BAV disease (59.4%), but CAE occurred similarly between subjects with (59.5%) and without (40.5%) ascending aortic pathology. The common appearance of CAE in patients with BAV could be independently confirmed in the BAV registry (38.9%), whereas CAE was found less frequently in family history positive MI patients (21.2%), and rarely in unrelated “real-life” catheterization patients (5.2%).
Conclusion: To our knowledge, our data show for the first time that ectatic coronary artery disease is a common appearance of BAV disease with and without ascending aortic aneurysm.
Article Information
vol. 130 no. Suppl 2 A19274
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Christine Meindl1;
- Birgit Achatz1;
- Deborah Huber1;
- Ute Hubauer1;
- Stefan Buchner1;
- Kurt Debl1;
- Sabine Fenk1;
- Christina Strack1;
- Christian Hengstenberg2;
- Heribert Schunkert2;
- Christa Meisinger3;
- Lars Maier1;
- Andrea Baessler1;
- Marcus Fischer1
- 1Internal Medicine II, Universitätsklinikum Regensburg, Regensburg, Germany
- 2Cardiology, Deutsches Herzzentrum, München, Germany
- 3Epidemiology, Helmholtz Zentrum, München, Germany
Abstract 17652: Application of Pediatric and Adult Lipid Treatment Guidelines to U.S. Adolescents Transitioning to Young Adulthood
Holly C Gooding, Angie M Rodday, John B Wong, Donald Lloyd-Jones, Matthew W Gillman, Laurel K Leslie, Sarah D de Ferranti
Circulation. 2014;130:A17652
Abstract
Introduction: Current pediatric and adult lipid treatment guidelines differ in their approach to pharmacologic treatment of cholesterol in adolescents and young adults. We hypothesized that a greater proportion of young people ages 17-21 would meet criteria for statin treatment under the pediatric guidelines compared to adult guidelines, but that overall eligibility for statin treatment would be low in this age group.
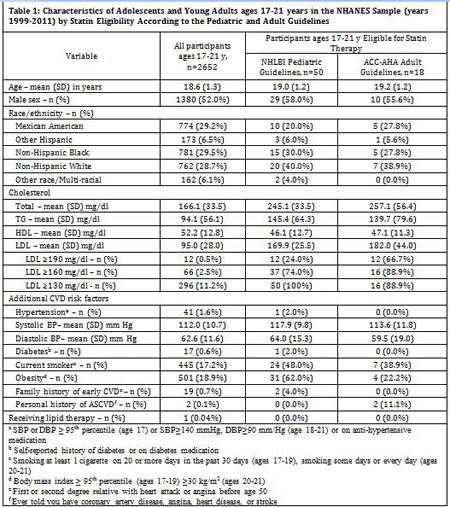
Methods: We applied treatment algorithms from the 2011 NHLBI Integrated Guidelines for Cardiovascular Health and Risk Reduction in Children and Adolescents and the 2013 ACC/AHA Guideline on the Treatment of Blood Cholesterol to Reduce Atherosclerotic Cardiovascular Risk in Adults to participants in the 1999-2011 National Health and Nutrition Examination Surveys who were 17-21 years of age and had an LDL level measured (n=2,652). We extrapolated the results to a population of 11.2 million individuals ages 17-21 years living in the US.
Results: Almost 2% of participants (n=50, 1.9%) qualified for statin treatment under the pediatric guidelines, but only 0.7% (n=18) met treatment criteria under the adult guidelines. Participants who met pediatric criteria had lower mean LDL levels but were more likely to have other cardiovascular risk factors, including hypertension, smoking, and obesity (Table 1). Despite the relatively low percentage of participants reaching LDL treatment thresholds under either guideline, 258,816 U.S. young people would be eligible for statin treatment under the pediatric guidelines and 84,651 would be eligible for treatment under the adult guidelines.
Conclusions: Providers who care for adolescents transitioning to adulthood are faced with incongruent lipid guidelines. Application of pediatric guidelines, which use a life course approach and consider additional cardiovascular risk factors beyond age, may result in statin treatment for more young people.
Article Information
vol. 130 no. Suppl 2 A17652
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Holly C Gooding1;
- Angie M Rodday2;
- John B Wong2;
- Donald Lloyd-Jones3;
- Matthew W Gillman4;
- Laurel K Leslie5;
- Sarah D de Ferranti1
- 1Pediatrics, Harvard Med Sch, Boston, MA
- 2Institute for Clinical Rsch and Health Policy Studies, Tufts Med Cntr, Boston, MA
- 3Dept of Preventive Medicine, Northwestern Univ Feinberg Sch of Medicine, Chicago, IL
- 4Population Medicine, Harvard Med Sch, Boston, MA
- 5Tufts Clinical and Translational Science Institute, Tufts Med Cntr, Boston, MA
Abstract 16262: Risk of Congenital Heart Surgery in Adults
Juergen Hoerer, Masamichi Ono, Jelena Kasnar-Samprec, Julie Cleuziou, Melchior Burri, Martina Strbad, Manfred Vogt, Rüdiger Lange, Christian Schreiber
Circulation. 2014;130:A16262
Abstract
Objective: There are currently no risk stratification models available for predicting the outcome following congenital heart surgery in adults. The Aristotle Basic Complexity (ABC), the Aristotle Comprehensive Complexity (ACC), the Risk Adjustment in Congenital Heart Surgery (RACHS-1), and the Society of Thoracic Surgeons (STS) – European Association for Cardiothoracic Surgery (EACTS) score are suitable for children. We sought to evaluate the predictive power of the ABC, ACC, RACHS-1, and STS-EACTS score for hospital mortality and complications after congenital heart surgery in adults.
Methods and results: Data of all consecutive patients aged 18 years or more, who underwent surgery for congenital heart disease between 2004 and 2014 at our institution, were collected. Complications were defined as reoperation, mechanical circulatory support, mechanical ventilatory support >24 hours, renal failure requiring dialysis, mediastinitis, persisting neurological deficit, and death during hospital admission or within 30 days. The discriminatory power of the scores was assessed using the area under the receiver operating characteristics (AuROC) curve.
846 operations were performed. Hospital mortality was 2.9%. Complications occurred in 15.6% of the patients. The prognostic significance of the ABC, ACC, RACHS-1, and STS-EACTS score for mortality was 0.67, 0.76, 0.60, and 0.74, respectively. The prognostic significance for complications was 0.65, 0.73, 0.60, and 0.70, respectively. Single ventricle physiology (p<0.001, OR=14.1) and older age (p=0.020, OR=1.04) were significant predictors for hospital mortality. Single ventricle physiology (p<0.001, OR=6.7), older age (p=0.003, OR=1.01), and male gender (p=0.022, OR=1.6) were significant predictors for complications.
Conclusions: The ACC score outperforms the ABC score since procedure dependent and independent factors are considered. The STS-EACTS score outperforms the RACHS-1 score since procedures can be categorized more precisely. The discriminative power of the ACC and the STS-EACTS score may be improved by including additional risk factors that are specifically present in the adult population.
Article Information
vol. 130 no. Suppl 2 A16262
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Juergen Hoerer1;
- Masamichi Ono1;
- Jelena Kasnar-Samprec1;
- Julie Cleuziou1;
- Melchior Burri1;
- Martina Strbad1;
- Manfred Vogt2;
- Rüdiger Lange1;
- Christian Schreiber1
- 1Dept of Cardiovascular Surgery, German Heart Cntr Munich, Munich, Germany
- 2Dept of Pediatric Cardiology and Congenital Heart Disease, German Heart Cntr Munich, Munich, Germany
Abstract 11608: Risk Factors for Atrial Arrhythmia and Thromboembolic Events in D-Transposition of the Great Arteries
Andrew Foy, Guodong Liu, William R Davidson, Douglas Leslie, Jennifer G Ting
Circulation. 2014;130:A11608
Abstract
Introduction: Atrial arrhythmia (AA) is an important problem in patients with adult congenital heart disease (ACHD). Relatively little is known about the prevalence and risk factors for thromboembolic events (TE) in patients with D-TGA and AA and further prospective study on this subpopulation of ACHD patients is difficult. Therefore, we sought to define the prevalence of AA and TE in a large, national sample of patients with D-transposition of the great arteries (D-TGA).
Methods: Retrospective analysis of health insurance claims data for a national sample of privately insured patients over the years 2008-2011. Individuals were included in the study cohort if they had a claim submitted for D-TGA at any time over the study period. The primary endpoints were claims submitted for AA, TE, and bleeding events; multivariate logistic regression was performed to identify independent variables associated with AA and TE.
Results: 3,414 patients were included in the final cohort, 253 (7.4%) had an AA, 38 (1.1%) had a TE, and 18 (0.5%) experienced a major bleeding event. The average age of patients with AA was 30.1 years compared to 15.6 years for those without AA. Independent variables associated with AA were age (OR 1.1, 95% CI:1.05-1.07), hypertension (HTN; OR 3.86, 95% CI:2.87-5.18), and hospitalization for congestive heart failure (CHF; OR 4.8, 95% CI:2.49-9.18). Those associated with TE were age (OR 1.06, 95% CI:1.04-1.09), and hospitalization for CHF (OR 5.87, 95% CI:2.01-16.58). AA was not associated with TE (OR 0.95, 95% CI:0.39-2.34).
Conclusions: AA is common in the D-TGA patient; AA is more common in the adult patient than in the pediatric patient. Heart failure is strongly associated with AA and TE; AA alone is not associated with TE. Bleeding events are not prevalent in this population. Despite use in CHADS-VASc for risk stratification of TE in adults with acquired heart disease, sex, diabetes, and HTN are not significantly associated with TE. These data further highlight important clinical differences and management strategies between pediatric, adult congenital heart disease and adult acquired heart disease patients.
Article Information
vol. 130 no. Suppl 2 A11608
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Program for Adult Congenital Heart Disease, Penn State Hershey Heart & Vascular Institute, Hershey, PA
- 2Public Health Sciences, Penn State College of Medicine, Hershey, PA
Abstract 20514: Adults With Hypoplastic Left Heart Syndrome: Outcomes in a New Cohort of Patients
William M Wilson, Candice Silversides, Anne Valente, Erwin Oechslin, S. Lucy Roche, Luke Burchill, Craig Broberg, Edward Hickey, Leeanne Grigg, Eliza Teo, Isabelle Von Der Muhll, Patrick Gibson, Jasmine Grewal, Paul Khairy, Matthias Greutmann, Kelsey Hickey, Yaso Emmanuel, Paul Clift, Rachel Wald
Circulation. 2014;130:A20514
Abstract
Background: Surgical palliation of hypoplastic left heart syndrome (HLHS) has only been possible for the past few decades. Prior to this, without heart transplantation, HLHS was universally fatal in infancy. The oldest survivors of palliated HLHS are only now entering adulthood and limited data are available regarding their welfare.
Methods: For this multi-center, cross-sectional, observational study, 6 adult congenital heart disease (ACHD) centers contributed data regarding all HLHS patients aged >18 years followed at their institution. HLHS was defined as a dominant right ventricle (RV) and diminutive left ventricle with a combination of mitral valve disease (stenosis [MS] or atresia [MA]) and aortic valve disease (stenosis [AS] or atresia [AA]). Patients with single RV physiology without hypoplasia of left heart inlet and outlet structures were excluded. All available clinical data, including cardiac imaging, cardiac catheterization results and exercise tests were reviewed.
Results: The study included 53 HLHS adults (65% male) with mean age 21.8±3.6 years. Underlying cardiac anatomy was AA&MA (n=21, 41%), AS&MS (n=19, 37%), AS&MA (n=10, 20%), and AA&MS (n=1, 2%). Stage 1 Norwood was completed at age 6.0±4.4 days, Glenn shunt at age 10.8±8.7 months and Fontan at age 3.3±2 years. Stage 1 cardiopulmonary bypass and circulatory arrest times were 94±46 and 59±2 minutes respectively. The mean duration of follow-up in an ACHD center was 3.4±2.5 years. After age 18 years, major adverse events had occurred in 15/53 patients (28%). These mutually exclusive events were: death (n=3, 6%), transplant (n=1, 2%), protein losing enteropathy (n=2, 4%), stroke (n=2, 4%), symptomatic ventricular tachycardia (n=1, 2%), heart failure requiring admission for intravenous therapy (n=3, 6%) and major cardiac surgery (n=3, 6% [aortic valve replacement n=1, tricuspid valve replacement n=2]).
Conclusions: Young adults with a Fontan palliation for HLHS are at high risk of major adverse cardiac events. It is essential that close attention is paid to successful transition of this vulnerable population into adult care and that these patients remain under vigilant specialist follow-up through adult life.
Article Information
vol. 130 no. Suppl 2 A20514
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- William M Wilson1;
- Candice Silversides1;
- Anne Valente2;
- Erwin Oechslin1;
- S. Lucy Roche1;
- Luke Burchill3;
- Craig Broberg3;
- Edward Hickey1;
- Leeanne Grigg4;
- Eliza Teo4;
- Isabelle Von Der Muhll5;
- Patrick Gibson5;
- Jasmine Grewal6;
- Paul Khairy7;
- Matthias Greutmann8;
- Kelsey Hickey2;
- Yaso Emmanuel9;
- Paul Clift9;
- Rachel Wald1
- 1Toronto Congenital Cardiac Cntr for Adults, Toronto General Hosp, Toronto, Canada
- 2Boston Adult Congenital Heart Program, Children’s Hosp Boston, Boston, MA
- 3Knight Cardiovascular Institute, Oregon Health and Science Univ, Portland, OR
- 4Dept of Cardiology, Royal Melbourne Hosp, Melbourne, Australia
- 5Mazakowski Alberta Heart Institute, Univ of Alberta, Edmonton, Canada
- 6Pacific Adult Congenital Heart Clinic, St Paul’s Hosp, Vancouver, Canada
- 7Adult Congenital Cntr, Montreal Heart Institute, Montreal, Canada
- 8Klinik fur Cardiologie, UniversitatsSpital Zurich, Zurich, Switzerland
- 9Dept of ACHD, Univ Hosps Birmingham, Birmingham, United Kingdom
Abstract 19314: Impact of Body Mass Index on Clinical Outcomes in Complex Adult Congenital Heart Disease
Norihisa Toh, Ines Uribe Morales, Zakariya Albinmousa, Tariq Saifullah, Rachael Hatton, Krishnakumar Nair, Rachel Wald, S. Lucy Roche
Circulation. 2014;130:A19314
Abstract
Background: Obesity can adversely affect most organ systems and increases the risk of comorbidities likely to be of consequence for patients with complex adult congenital heart disease (ACHD). Conversely, several studies have demonstrated that low body mass index (BMI) is a risk factor for heart failure and adverse outcomes after cardiac surgery. However, there are currently no data regarding the impact of BMI in ACHD.
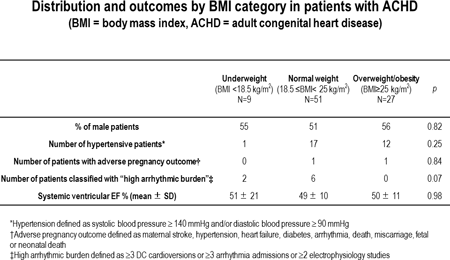
Methods: We examined the charts of 87 randomly selected, complex ACHD patients whose first visit to our institution was at 18-22 years old. Patients were categorized according to BMI at initial visit: underweight (BMI < 18.5 kg/m2), normal (BMI 18.5 – 24.9 kg/m2), overweight/obese (BMI ≥ 25 kg/m2). Events occurring during follow-up were recorded. Data was censured on 1/1/2014. Cardiac events were defined as a composite of cardiac death, heart transplantation or admission for heart failure.
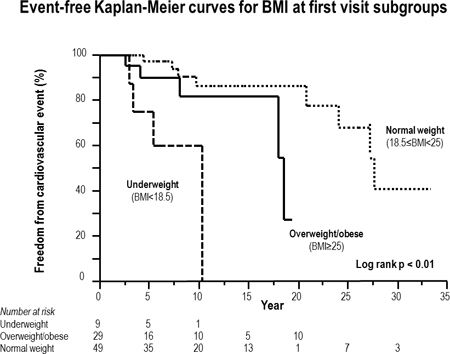
Results: The cohort included patients with the following diagnoses: tetralogy of Fallot n=31, Mustard n=28, Fontan n=17, ccTGA n=9 and aortic coarctation n=2. The median (IQR) duration of follow-up was 8.7 (4.2 – 1.8) years. See table for distribution and outcomes by BMI category. Cardiac events occurred in 17/87 patients. After adjustment for age, sex, and underlying disease, the underweight group had increased risk of cardiac events (HR=12.9, 95% CI: 2.8-61.5, p < 0.05). Kaplan-Meier curves demonstrate the poorer prognosis of underweight patients (Figure).
Conclusions: Underweight was associated with increased risk of late cardiac events in ACHD patients. We were unable to demonstrate significant overweight/obesity impact.
Article Information
vol. 130 no. Suppl 2 A19314
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Norihisa Toh;
- Ines Uribe Morales;
- Zakariya Albinmousa;
- Tariq Saifullah;
- Rachael Hatton;
- Krishnakumar Nair;
- Rachel Wald;
- S. Lucy Roche
- Div of Cardiology, Toronto Congenital Cardiac Cntr for Adults, Univ of Toronto, Toronto, Canada
Abstract 18829: Economic Self-sufficiency and Educational Attainment is Worse in Adult Congenital Heart Disease Survivors
Nicolas L Madsen, Bradley S Marino, Jessica G Woo, Susie Antonsen, Jorgen Videbaek, Morten S Olsen
Circulation. 2014;130:A18829
Abstract
Introduction: Among congenital heart disease (CHD) survivors, there is a distinct pattern of neurodevelopmental and behavioral impairment.
Objective: To compare attainment of self-sufficiency and its interaction with educational achievement in adult CHD survivors with sibling and general population controls.
Methods: Using Danish population-based registries this cohort study aimed to include all CHD survivors greater than 13 years born between 1963-1993. Comparison cohorts included: 1) A sibling cohort, and 2) A population cohort matched (1:10) on birth year and gender. We computed cumulative incidences of time to first full year of economic self-sufficiency, as well as completed vocational, high school, and higher education. Self-sufficiency was defined by Statistics Denmark standard; total income from all available sources greater than 50% of a student’s annual subsidy.
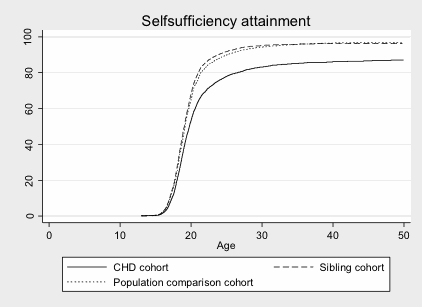
Results: We identified 10,259 CHD survivors, 8,634 full siblings and 101,653 population controls. The cumulative incidence of self-sufficiency at age 20 years for CHD patients (53.2%) was significantly lower than sibling (67.9%) and population controls (64.7%) (p<0.05 for both comparisons). By age 40, CHD survivors remained less self-sufficient (85.7%) than both comparison cohorts (sibling 96.3% and population 96.6%, p<0.05) (Figure). By age 30, CHD survivors were significantly less likely to attain vocational, high school, or higher education (9.9%, 26.6%, and 22.8%, respectively) than their siblings (11.4%, 31.1%, and 25.3%, p<0.05 for each comparison). Among those achieving these educational milestones, differences in self-sufficiency between CHD survivors and controls became non-significant by age 40.
Conclusion: CHD is associated with less economic self-sufficiency. This is not explained by differences in socio-economic status as demonstrated by comparison to sibling controls. This data suggests that educationally targeted therapies may improve self-sufficiency.
Article Information
vol. 130 no. Suppl 2 A18829
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Nicolas L Madsen1;
- Bradley S Marino2;
- Jessica G Woo1;
- Susie Antonsen3;
- Jorgen Videbaek3;
- Morten S Olsen3
- 1Pediatrics, Cincinnati Children’s Hosp Med Cntr, Cincinnati, OH
- 2Pediatrics, Ann & Robert H. Lurie Children’s Hosp of Chicago, Chicago, IL
- 3Clinical Epidemiology, Univ of Aarhus, Aarhus, Denmark
Abstract 18929: Pacing in the Adult Fontan Patient: Risky Business?
Matthew J Lewis, Nalini Colaco, Jonathan Ginns, Marlon Rosenbaum
Circulation. 2014;130:A18929
Abstract
Introduction: Chronic ventricular pacing can induce a cardiomyopathy in patients with a biventricular heart; however, the effect of chronic pacing in adult patients with a Fontan has not been well characterized. We hypothesized that paced adult Fontan patients would be at higher risk for death or heart transplant.
Methods: We performed a retrospective, cohort of study of all adult Fontan patients at the Schneeweiss Adult Congenital Heart Center seen between 1/1997 and 5/2014. Two Cohorts were defined based on whether a patient did or did not have a permanent pacemaker. Demographic and clinical characteristics were collected via chart review. The primary endpoint was a composite of death or heart transplant.
Results: Of the 98 adult Fontan patients followed (mean age at last follow-up 32± 8 years), 30 (31%) had a pacemaker. Pacemaker specific data was available on 25 of the 30 (83%) paced patients. Of those, 88% were paced >50% of the time. Patient diagnoses included double inlet left ventricle in 33 (34%), tricuspid atresia in 26 (27%), hypoplastic left heart in 9 (9%), heterotaxy in 8 (8%), and 22 (22%) with other diagnoses. Fifty-two patients (53%) had a classic RA-PA Fontan and 46 (47%) had a lateral tunnel or extracardiac Fontan.
Over the study period, 16 patients met the primary endpoint and 12 (75%) were paced. Paced patients were significantly more likely to have worse functional status (p<0.001), be on diuretics (p<0.001), and have a higher mean creatinine (P=0.025), mean total bilirubin (p=0.025), and mean Fontan pressure (p<0.001). Pacing was associated with >4-fold increase in the rate of death or heart transplant (p=0.009) in a multivariate cox-proportional hazard model that included Fontan type, age at Fontan completion, age at follow-up, and pacing status.
Discussion: In our cohort of 98 adult Fontan patients, paced patients were more likely to have worse functional status, require diuretics and had a >4-fold increased risk of death or heart transplant. These results suggest that chronic pacing may be detrimental in this population.
Article Information
vol. 130 no. Suppl 2 A18929
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Cardiology, Columbia Univ/New York Presbyterian Hosp, New York, NY
- 2Medicine, Columbia Univ/New York Presbyterian Hosp, New York, NY
Abstract 13187: Abnormalities in Cardiac Strain are Found in Young Adults with Type 2 Diabetes Mellitus
Nicolas L Madsen, Philip R Khoury, Zhiqian Gao, Stephanie N Stewart, Lauren Longshore, Amy Shah, Lawrence M Dolan, Thomas R Kimball, Elaine M Urbina
Circulation. 2014;130:A13187
Abstract
Introduction: Diabetic cardiomyopathy in adults begins with diastolic dysfunction progressing to systolic dysfunction and heart failure. Tissue Doppler imaging to measure strain and strain rate may demonstrate abnormalities earlier in the process than traditional echo. We hypothesized that abnormal strain can be found in young adults with Type II Diabetes Mellitus (T2DM).
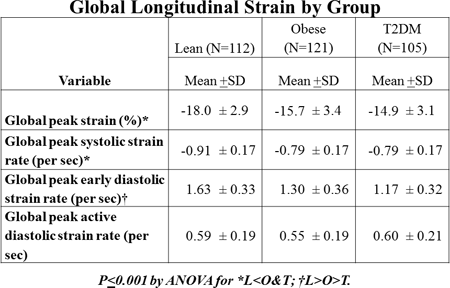
Methods: Comprehensive echocardiography and speckle-tracking imaging was measured in 338 subjects (22.2 + 3.7 years, 38% male, 63% African American) stratified into lean (L=112), obese (O=121) and T2DM (T=105) groups. Anthropometrics, BP, HR, fasting lipids, glucose and CRP were collected. Echocardiograms were performed on a GE Vivid 7 machine and read with EchoPAC®software for global longitudinal (4-chamber) strain (GS) and strain rate in systole (GSRs), early diastole (GSRe) and active diastolic phase (GSRa). ANOVA was performed to compare differences among groups for CV risk factors and strain measures and ANCOVA to determine if presence of T2DM remained an independent predictor of strain after correction for risk factors (full model=age, sex, race, group (L,O,T), MAP, HR and lab values).
Results: Lean subjects were lighter with lower BP, lipids, CRP and glucose. Lean subjects had superior cardiac function with more negative GS and GSRs and more positive GSRe than O & T (p<0.001). There was no group difference in GSRa. Major determinants of global strain were sex, adiposity and MAP with age, HR, HDL contributing for GS, HDL and T2DM for GSRse and HDL & glucose for GSRa (R2: GS=0.38, GSRs=0.24, GSRe=0.41, GSRa=0.13). Presence of T2DM remained an independent predictor of GSRe even after correction for CV risk factors.
Conclusions: Obesity and BP influence strain in young adults. Presence of T2DM is associated with early diastolic strain abnormalities beyond that predicted by CV risk factors alone. Control of obesity and T2DM is needed in young adults to prevent the risk of future heart failure.
Article Information
vol. 130 no. Suppl 2 A13187
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Nicolas L Madsen;
- Philip R Khoury;
- Zhiqian Gao;
- Stephanie N Stewart;
- Lauren Longshore;
- Amy Shah;
- Lawrence M Dolan;
- Thomas R Kimball;
- Elaine M Urbina
- Cardiology, Cincinnati Childrens Hosp, Cincinnati, OH
Abstract 13223: Structural Equation Modeling Demonstrates only an Indirect Effect of Obesity on Carotid Intima-Media thickness in Adolescents and Young Adults
Zhiqian Gao, Philip R Khoury, Connie E McCoy, Amy S Shah, Thomas R Kimball, Lawrence M Dolan, Elaine M Urbina
Circulation. 2014;130:A13223
Abstract
Introduction: Increased carotid intima-media thickness (cIMT) is associated with cardiovascular (CV) events in adults. Thicker cIMT is found in youth with elevated CV risk factors including obesity although biologically plausible reasons for a direct effect are not clear. We hypothesized that obesity affects cIMT only indirectly through other CV risk factors and this could be demonstrated with use of structural equation modeling (SEM).
Methods: Ultrasound of the right and left common, bulb and internal carotid arteries was performed in 784 adolescent and young adults (10-24 years old, 65% female, 41% Caucasian, 32% T2DM). Demographics, anthropometrics and fasting laboratory data were collected. Traditional multiple regression analyses (MRA) were performed to assess independent determinants of cIMT. Analyses were then repeated with SEM.
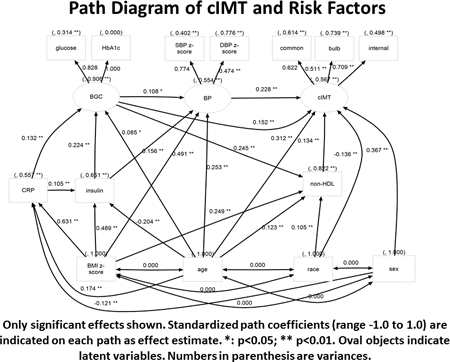
Results: MRA models explained 11%-22% of variation of common, bulb and internal cIMT. Obesity, age, sex and SBP z-score were significant determinants of all cIMT segments. Race, presence of T2DM, HbA1c and non-HDL contributed for some segments. In SEM, latent variable “cIMT” was used to represent the 3 segments of cIMT. Latent variable “BP” was extracted from SBP and DBP z-score. Latent variable “BGC” (blood glucose control) was extracted from fasting glucose and HbA1c. The final SEM explained a larger amount of the variance of cIMT (43%). The largest direct effect on cIMT was age followed by BP, blood glucose control and non-HDL. BMI, another central risk factor in the pathway towards atherosclerosis, only had a significant indirect effect through blood glucose control, BP & non-HDL. CRP had a small indirect effect through blood glucose control. We conclude that traditional CV risk factors have important direct effects on cIMT in adolescents and young adults but adiposity exerts its influence only through other CV risk factors. SEM may be a useful tool in modeling complex biological pathways.
Article Information
vol. 130 no. Suppl 2 A13223
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Zhiqian Gao;
- Philip R Khoury;
- Connie E McCoy;
- Amy S Shah;
- Thomas R Kimball;
- Lawrence M Dolan;
- Elaine M Urbina
- Cardiology, Cincinnati Childrens Hosp, Cincinnati, OH
Abstract 12649: Cardiovascular Exercise Reduces Anxiety Symptoms in Adults With Congenital Heart Disease
Munziba Khan, Megan Smith, Vicki Freedenberg, Nancy Klein, Anitha S John
Circulation. 2014;130:A12649
Abstract
Background: Adult congenital heart disease (ACHD) patients (pts) have high rates of untreated depression and anxiety disorders. We evaluated the association between self-reported depression and anxiety symptoms and cardiopulmonary exercise.
Methods: From 2009 to 2013, 193 ACHD pts (46% male) completed clinical questionnaires including data regarding symptoms of depression and anxiety, and frequency of cardiopulmonary exercise. Data were collected by retrospective review.
Results: Mean age was 31 + 10 years. Disease severity was classified as: mild (20%), moderate (48%), and severe (32%). Nineteen percent of pts reported being depressed often and 26% were nervous or anxious. There was no association between age, gender or severity of disease and depression or anxiety symptoms.
Exercise frequency was classified as none (27%), low (<3x/month, 6%), occasional (<2x/week, 8%) or frequent (> 2x/week, 58%). There was no significant association between severity of disease and frequency of exercise. Fewer pts who exercised reported anxiety symptoms compared to those who did not exercise (21% vs 35%, p=0.04). When adjusted for age, gender and severity of illness, pts who exercised frequently were half as likely to report symptoms of anxiety (OR 0.46; 95% CI 0.23 to 0.91) as those who never exercised.
Exercise stress test data was available in 85 pts. Frequent exercisers had higher peak VO2 (28.6 + 7.8 versus 24.9 + 6.8 ml/kg/min, p=0.03), predicted VO2 (81.4% + 19.6 vs 66.9% + 14.9, p=0.001), and maximal METS (8.3 + 2.2 vs 7.0 + 2, p=0.01). Frequent exercisers also had lower resting heart rates (72 + 13bpm vs 79 + 12 bpm, p=0.02). Disease severity in pts who exercised frequently was: mild (23%), moderate (54%), and severe (23%).
Fourteen percent of pts were on antidepressant/antianxiety (AD/AA) medications (meds); 56% of this subgroup still reported anxiety symptoms. There was a greater percentage of non-exercisers vs frequent exercisers in those with continued symptoms (50% vs 14%).
Conclusion: Regular cardiopulmonary exercise by ACHD pts is associated with decreased self-reported anxiety symptoms and improved exercise capacity. Cardiopulmonary exercise may be an adjunct mode of treatment for anxiety disorder, but further investigation is needed.
Article Information
vol. 130 no. Suppl 2 A12649
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Munziba Khan;
- Megan Smith;
- Vicki Freedenberg;
- Nancy Klein;
- Anitha S John
- Cardiology, Children’s National Med Cntr, Washington, DC
Abstract 12028: Left Ventricular Contractile Reserve Assessed by Exercise Stress Echocardiogram in Adults With Repaired Tetralogy of Fallot: A Novel Early Marker of Intrinsic Myocardial Disease?
Amelie Michaud, Pier-Anne Gilbert, Michael A Gatzoulis, Philippe Pibarot, Josep Rodés-Cabau, Jean Perron, Elisabeth Bedard
Circulation. 2014;130:A12028
Abstract
Background and objective: Left (LV) and right (RV) ventricular systolic dysfunction have been identified as risk factors for adverse outcomes in patients with repaired Tetralogy of Fallot (TOF) and early detection is mandatory. Using exercice stress echocardiography (ESE), this study investigated LV contractile reserve in TOF patients compared with healthy adults.
Methods: 28 adults with repaired TOF and 28 age- and sex- matched controls were propectively evaluated with supine bicycle ESE. Contractile reserve was evaluated by mesuring LV stroke volume at rest and on exertion.
Results: TOF patients (age, 35±10 years; male, 14) had a smaller increase in LV stroke volume on maximal exertion than controls, even when corrected for maximum workload performed (12±16% vs 28±16%, p=0,015) (Figure 1). No correlation was found between exercice stroke volume and degree of pulmonary insufficiency or RV and LV size and function at rest.
Conclusions: LV contractile reserve is impaired in patients with repaired TOF, independently of their degree of pulmonary insufficiency or RV and LV size and function. Importantly, these findings may suggest an intrinsic LV myocardial abnormality in TOF patients, for which early detection may be achieved by ESE. Larger studies are needed to determine if impaired contractile reserve represents an independent risk factor for ventricular tachycardia and or death in these patients.
Article Information
vol. 130 no. Suppl 2 A12028
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Amelie Michaud1;
- Pier-Anne Gilbert1;
- Michael A Gatzoulis2;
- Philippe Pibarot1;
- Josep Rodés-Cabau1;
- Jean Perron3;
- Elisabeth Bedard1
- 1Cardiology, Quebec Heart and Lung Institute, Quebec, Canada
- 2Cardiology, Royal Brompton Hosp, London, United Kingdom
- 3Cardiac Surgery, Quebec Heart and Lung Institute, Quebec, Canada
Abstract 12140: Prognostic Value of Multiple Biomarkers on Mortality in Adults Congenital Heart Disease: Comparison of Single- and Two-Ventricle Physiology
Kenji Miyamoto, Daiji Takeuchi, Kei Inai, Tokuko Shinohara, Toshio Nakanishi
Circulation. 2014;130:A12140
Abstract
Introduction: Although many biomarkers are associated with heart failure, limited data is available on their prognostic predictive value in adults with congenital heart disease (ACHD). We investigate the potential of various biomarkers to predict ACHD mortality.
Methods and Results: This is a single-center, retrospective cohort study. Blood levels of neurohormones (endothelin-1 [ET1], norepinephrine [NE], aldosterone, and plasma renin activity [PRA]); inflammatory biomarkers (high sensitivity C-reactive protein [hs-CRP], high sensitivity tumor necrosis factor [hs-TNF]-α, soluble TNF receptor type I and II [sTNF-RI and sTNF-RII], and interleukin-6 [IL-6]); and brain natriuretic peptide (BNP) were measured in 115 ACHD (mean age, 30 ± 10 years). NYHA class was I/ II in 86% and III/IV in 14%. The subjects were divided into two groups: patients with single-ventricle (SV group, n = 65) and with two-ventricle physiology (TV group, n = 50). We retrospectively analyzed the relationship between levels of biomarkers and cardiovascular death. During a mean follow-up period of 4.6 years, 17 (14%) cardiovascular deaths occurred, including 7 in the SV group. Univariate cox regression analysis in all subjects showed strong association between elevated levels of ET1, NE, RPA, hs-CRP, sTNF-RI and II, IL-6, and BNP and cardiovascular death (p< 0.05). In the SV group, using multivariate cox regression model, BNP and sTNF-RI were the most powerful predictors in these biomarkers (adjusted hazard ratio [aHR] of BNP: 14.84; 95%CI: 2.21-99.36 per 1 SD increase, p = 0.005) (aHR of sTNF-RI: 2.30; 95%CI: 1.91-4.55 per 1 SD increase, p = 0.017). The optimal cut-off values of BNP and sTNF-RI for mortality were 196 pg/mL and 1.26 ng/mL, respectively. Conversely, in the TV group, only IL-6 was an independent predictor of mortality (aHR: 3.24; 95%CI: 1.57-6.68 per 1 SD increase, p = 0.001), while BNP was not strongly associated with outcomes. The optimal cut-off value of IL-6 for mortality was 2.3 pg/mL.
Conclusion: Various biomarkers, including sTNF-R, BNP, and IL-6, are associated with prognosis in overall ACHD. The most prominent mortality predictors might differ, due to differences in SV or TV physiology.
Article Information
vol. 130 no. Suppl 2 A12140
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Kenji Miyamoto;
- Daiji Takeuchi;
- Kei Inai;
- Tokuko Shinohara;
- Toshio Nakanishi
- Dept of Pediatric Cardiology, Towkyo Women’s Med Univ, Tokyo, Japan
Abstract 19820: Heterogeneity of Disease Progression Rates in Patients With Bicuspid Aortic Valve: Is There a Suitable Bio-marker?
Thanh H Nguyen, Ranjit Shah, Matthew Chapman, Onn A Ali, John D Horowitz
Circulation. 2014;130:A19820
Abstract
Introduction: Bicuspid aortic valve (BAV) is associated with inflammatory activation and endothelial dysfunction. Worsening of aortic valve stenosis/regurgitation represents the most common complication of BAV, but clinical and biochemical markers of disease progression are not currently available.
Objective: The objective of the current study was to (1) evaluate rates of progression of aortopathy and valve dysfunction, and (2) identify correlates of accelerated progression among BAV patients.
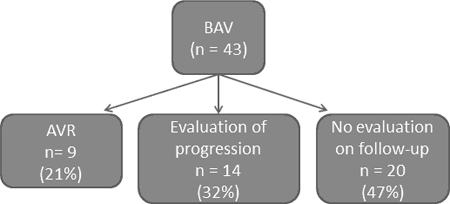
Methods: 43 BAV patients aged 45 ± 16 (SD) years were evaluated clinically and with echo/MRI. Inflammatory activation was assessed via hs-CRP and myeloperoxidase (MPO) levels, endothelial function/NO responsiveness via flow-mediated dilatation (FMD), plasma asymmetric dimethylarginine (ADMA) levels, inhibition of platelet aggregation by SNP, and endothelial progenitor cell counts. Follow-up was undertaken after 47 ±16 months to determine rate of progression and of need for valve replacement; some patients did not undergo repeat echo (Figure). Biochemical/physiological correlates of valve dysfunction and aortic dimensions were sought via univariate and multiple linear regression analyses.
Results: AV max increased from 2.1 ± 0.6 to 2.5 ± 1.1(m/s), p < 0.05, while ascending aorta (Asc Ao) dimensions increased from 18.2 ± 6.1 to 21.2 ± 4.9 (mm/m2), p < 0.05 over the monitoring period. Although there was a direct correlation (p < 0.01) between baseline AV max and MPO levels on multivariate analysis, this correlation did not extend to progression rates. Indeed neither clinical nor biochemical parameters predicted accelerated progression.
Conclusions: (1) Progression of valve disease and aortopathy in BAV is often rapid, with annual mean rate of 0.1 m/s for AV max and 0.4 mm/m2 for Asc Ao. (2) Despite physiological evidence linking BAV with inflammation and endothelial dysfunction, these biochemical factors did not appear to predict rapid disease progression.
Article Information
vol. 130 no. Suppl 2 A19820
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Cardiology, The Queen Elizabeth Hosp, The Univ of Adelaide, Adelaide, Australia
- 2Cardiology, The Queen Elizabeth Hosp, Adelaide, Australia
Abstract 19355: Lower Heart Rates are Associated with Better Mid-term Outcomes in Fontan Patients
Clara Kurishima, Hideaki Senzaki, Tokuko Shinohara, Toshio Nakanishi
Circulation. 2014;130:A19355
Abstract
Background: In adult heart failure, higher heart rates (HR) are known to be associated with increased risks of myocardial infarction and sudden cardiac death. Thus, HR modulation has been increasingly recognized as a potentially effective therapy for heart failure. HR may also be a useful therapeutic target in patients after the Fontan surgery, in which effective treatment to improve prognosis remains to be established. We hypothesized that lower HR is associated with better mid-term outcomes in Fontan patients.
Methods: We retrospectively analyzed 24-hour Holter ECG in 56 consecutive patients in the chronic phase (at least 1 year) after the Fontan operation and in the sinus rhythm (mean age; 19 years, range; 9-49 years). Data for minimum, maximum, and average HR were extracted. We then examined the relationships between HR values and mid-term hemodynamic and clinical (6-min walk-distance) outcomes. Mid-term outcomes were assessed both at 1-3 years (mean 1.5 years) after the initial Holter recording (Group 1) and at more than 3 years (mean 4.9 years) after the initial Holter recording (Group 2).
Results: Lower values of mean and minimum HR were significantly correlated with lower CVP in both groups (P<0.05, for each). Mean and minimum HR were also significantly and negatively correlated with the 6-min walk-distance in Group 2 (P <0.05 for each). The results were similar after controlling for age by multivariate analysis. Importantly, lower HR was not associated with decreased cardiac output.
Conclusions: The present study demonstrated for the first time that lower HR can lead to better mid-term outcomes of hemodynamics and exercise capacity in Fontan patients. HR can be an important therapeutic target to improve the prognosis after the Fontan operation.
Article Information
vol. 130 no. Suppl 2 A19355
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Pediatric Cardiology, Tokyo Women’s Med Univ, Tokyo, Japan
- 2Pediatric Cardiology, Saitama Med Cntr, Saitama Med Univ, Saitama, Japan
Abstract 18937: Ebstein’s Anomaly in Adults – Favourable Outcomes from a Single Specialist Centre Experience
Preeti Choudhary, Queenie Luu, Carla Caniffe, Dan Jackson, David S Celermajer
Circulation. 2014;130:A18937
Abstract
Introduction: Ebstein’s anomaly (EA) accounts for less than 1% of all congenital heart disease (CHD) and outcome data in adults is scarce. In our adult CHD centre, we have practised an uniform approach of “watchful waiting” with surgery reserved for either those with refractory arrhythmia or worsening dyspnoea, with lower threshold for cases with tricuspid valves amenable to repair (rather than require replacement), especially with adequate biventricular volumes. We aimed to evaluate the long-term outcomes of our EA patients managed with this approach.
Methods: All patients with EA and normal cardiac connections reviewed at least once in the adult CHD clinic were included. Outcome data was ascertained by reviewing the National Deaths registry and medical records.
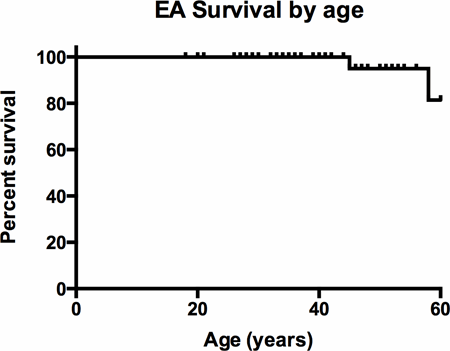
Results: 51 EA patients (17 males, median age at diagnosis 13 years (0-63), mean age at first adult CHD clinic review 33+/-13 years) were followed for a mean period of 21+/-14 years. Of these, 18 patients (35%) had supraventricular arrhythmia with Wolff-Parkinson-White syndrome in 5 patients (10%). 16 patients (31%) underwent catheter ablation with sustained freedom from arrhythmia in 9 (56%) patients. Five patients (10%) required pacemaker implantation. 16 patients (31%) underwent tricuspid valve (TV) surgery (9 repair and 7 replacement). 7 patients had anti-arrhythmia surgery at the time of TV surgery, with re-do surgery required in 3. Small LV (defined as end-diastolic diameter <40mm in adulthood) was present in 20 patients. There were three deaths; a 58 y.o. female with cardiogenic shock after third time sternotomy and valve surgery, a 45 y.o. female with progressive heart failure and a 72 y.o. male with mild EA and unknown cause of death. Kaplan-Meier analysis showed 100% survival to age 40, 95% to age 50 and 81% to age 60.
Conclusion: EA in adulthood often has severe morphological abnormalities but with a conservative management strategy, we demonstrate good medium-term survival.
Article Information
vol. 130 no. Suppl 2 A18937
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Preeti Choudhary;
- Queenie Luu;
- Carla Caniffe;
- Dan Jackson;
- David S Celermajer
- Cardiology, Univ of Sydney, Royal Prince Alfred Hosp, Sydney, Australia
Abstract 18723: Relationship of Health-Related Quality of Life to Neurodevelopmental Function in Fontan Adolescents
Carolyn Dunbar-Masterson, Christian Stopp, David C Bellinger, Dana L Bernson, David R DeMaso, Michael J Rivkin, Jane W Newburger
Circulation. 2014;130:A18723
Abstract
Background: Health-related quality of life (QOL) in Fontan patients has been shown to be related to patient and medical characteristics as well as to parent-reported problems with learning and behavior. However, no studies have examined the relationships of QOL to specific neurodevelopmental (ND) domains measured with concurrent in-person testing during adolescence.
Hypothesis: Worse QOL will be related to specific ND disabilities.
Methods: Parents of 152 Fontan patients (14.5±2.9y) and 107 local referent subjects (15.3±1.8y) completed the Child Health Questionnaire, generating summary scores for psychosocial (PsS) and physical (PhS) functioning. Subjects also underwent concurrent in-person testing with a battery of ND tests.
Results: Fontan patients, compared to referents, had lower PsS scores (48.2±11.1 vs. 57.1±4.4, P<0.001) and PhS scores (45.3±11.1 vs. 56.0±4.5, P<0.001). Even those without known genetic abnormality had lower PsS and PhS scores than referents (P<0.001 each). Lower PsS scores were highly associated with worse executive function (Behavior Rating Inventory of Executive Function, parent, r=-0.71, P<0.001) and attention (Conners ADHD T-score, parent, r=-0.68, P<0.001) after adjusting for concurrent social status, genetic abnormality, and previous Norwood procedure. Lower PsS scores were also related to lower measures of intelligence (Wechsler Intelligence Scale, Full-Scale IQ, r=0.25, P=0.002), achievement (Wechsler Individual Achievement Test, math, r=0.24, P=0.003), memory (Children’s Memory Scale, r=0.37, P<0.001; Wechsler’s Memory Scale, composite, r=0.38, p=0.03), and visual-spatial skills (Test of Visual-Perceptual Skills, composite, r=0.22, P=0.04) as well as to greater indices of depression (Child Depression Inventory Total Score, r=-0.39, P<0.001), anxiety (Revised Children’s Manifest Anxiety Scale, r=-0.29, P<0.001) and autism (Autism Spectrum Quotient, r=-0.31, P<0.001). Lower PhS scores were associated with few ND outcomes.
Conclusions: Worse psychosocial health in Fontan adolescents is highly related to worse ND performance. The strong correlations of worse psychosocial health with executive dysfunction and ADHD suggest the importance of interventions targeted to these domains.
Article Information
vol. 130 no. Suppl 2 A18723
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Carolyn Dunbar-Masterson1;
- Christian Stopp1;
- David C Bellinger2;
- Dana L Bernson1;
- David R DeMaso3;
- Michael J Rivkin2;
- Jane W Newburger1
- 1Cardiology, Boston Children’s Hosp, Boston, MA
- 2Neurology, Boston Children’s Hosp, Boston, MA
- 3Psychiatry, Boston Children’s Hosp, Boston, MA
Abstract 18614: Long-term Psychosocial Outcomes of Congenital Heart Disease: Neurocognitive Performance, Quality of Life, Psychosocial Adjustment and Psychiatric Morbidity of Adolescents and Young Adults Surviving Their Disease
Maria Emilia G Areias, Stefanie Melo, João Pedro Lopes, Filipa Rodrigues, Ana Catarina Nascimento, Daniela Cerqueira, Liliana Gomes, Anabela Estrela, Joana Miranda, Filipa Vilacova, Cláudia Moura, Joana Soares, Bruno Peixoto, Jorge Quintas, José Carlos Areias
Circulation. 2014;130:A18614
Abstract
Objectives: to study neurocognitive performance (NP) of CHD patients and to determine whether is related to parameters of fetal development registered at birth, head circumference (HC), weight (W) and length (L) and neonatal parameters (APGAR 1, 5); to study their quality of life (QOL), psychiatric morbidity (PM), psychosocial adjustment (PSA) and traits of personality (TP).
Methods: 266 CHD patients, 148 male, aged from 12 to 30 years (mean= 18.00 ± 3.22), 103 cyanotic, and 119 healthy controls (56 males, mean=18.41±3.20) participated. Clinical data were collected. Neuropsychological assessment included Wechsler’s Digit Test (direct and reverse) and Symbol Search, Rey’s Complex Figure, BADS’s Key Searching Test, Color-Word Stroop Test, Trail Making Test (A, B) and Logical Memory Task. Participants were interviewed on social support, family educational style, self-image, physical limitations, completed a psychiatric interview (SADS-L) and self-report questionnaires on QOL (WHOQOL-BREF), PSA (YSR and ASR) and TP (NEOPI-R). HC, W and L and APGAR were collected.
Results: CHD patients had a significantly worse NP than healthy controls in all tests, and the cyanotic worse than the acyanotic patients (but not significantly). Several correlations were apparent between fetal parameters (HC, W and L) and neuropsychological abilities in CHD. However, low weight at birth, cyanosis and male gender are the main predictors of bad NP later on in CHD patients (R=0.414; R2=0.171; F=5.787; p=0.001; β=1.654; t=2.858; p=0.005; β=1.881; t=2.377; p=0.020; b=1.624; t=2.062; p=0.042). We found a 15.3% lifetime prevalence of psychopathology (18.5% in females). Comparing to normal population, our patients have better QOL in environmental (t=6.907; p=0.000), social relationships (t=5.102; p=0.000) and general dimensions (t=2.558; p=0.011). Complex CHD reported worse QOL in physical dimension (U=3576.500; p=0.001) than those with moderate/mild forms of disease; Female patients showed worse PSA, with more withdrawal, anxiety/depression and internalization.
Conclusion: CHD patients have worse NP than controls; low weight at birth, male gender and the presence of cyanosis predict bad NP in CHD patients; patients seem to be more prone to PM, worse PSA and SP.
Article Information
vol. 130 no. Suppl 2 A18614
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Maria Emilia G Areias1;
- Stefanie Melo1;
- João Pedro Lopes1;
- Filipa Rodrigues1;
- Ana Catarina Nascimento1;
- Daniela Cerqueira1;
- Liliana Gomes1;
- Anabela Estrela1;
- Joana Miranda2;
- Filipa Vilacova2;
- Cláudia Moura2;
- Joana Soares1;
- Bruno Peixoto1;
- Jorge Quintas3;
- José Carlos Areias2
- 1Psychology, Instituto Superior de Ciências da Saúde – Norte (CESPU), Gandra-Paredes, Portugal
- 2Pediatric Cardiology, Hosp S. João, Porto, Porto, Portugal
- 3Criminology, Faculty of Law, Porto, Portugal
Abstract 18452: Outcome of Pregnancy in Women with Aortic Disease: Data from ROPAC
Iris M van Hagen, Mark R Johnson, Roger Hall, Jolien W Roos-Hesselink
Circulation. 2014;130:A18452
Abstract
Objectives: Cardiovascular disease is one of the major causes of maternal mortality. Pregnancy induces marked hemodynamic changes and may weaken the structure of the vessel wall. The aim of the present study is to describe the outcome of pregnancy in patients with aortic disease.
Methods and Results: In the Registry Of Pregnancy And Cardiac disease (ROPAC), 2966 patients were enrolled from 39 countries. One hundred and one patients had aortic disease: 56 patients with Marfan syndrome (55.4%), 12 associated with bicuspid aortic valve (11.9%), 2 with Turner Syndrome (2.0%), 2 with familial thoracic aortic aneurysm disease (2.0%) and 29 with other aortic diseases (28.7%) e.g. Takayasu arteritis or previous aortic dissection without specified diagnosis. During pregnancy, aortic dissection occurred in 3 patients: a type B dissection in a Marfan patient at 25 weeks with a prior aortic diameter of 42 mm; a type A dissection in a Marfan patient at 37 weeks (pre-pregnancy aortic diameter not reported) and at 26 weeks in one patient with a prior aneurysm (diameter 60mm). The first two patients used a beta-blocker during pregnancy. All patients underwent surgical replacement of the aorta directly after Caesarean Section (CS). The patient with type B dissection had her caesarean section at 33 weeks. No fetal loss occurred. A fourth patient, also affected with Marfan syndrome, suffered a type A aortic dissection at 7 days after delivery. Her pre-pregnancy aortic diameter was 26mm and she did not use a beta-blocker during pregnancy. There was no maternal mortality and only one miscarriage in all 101 cases. CS was performed in 64.6% of the patients compared with 47.1% (p=0.001) in the rest of the registry.
Conclusion: Pregnant patients with aortic disease are at risk of aortic dissection with an incidence in Marfan patients of 5.4%. This is mainly seen in the second half of pregnancy or shortly after delivery. In this registry, no maternal mortality or fetal loss were observed after aortic dissection.
Article Information
vol. 130 no. Suppl 2 A18452
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Cardiology, Erasmus MC, Rotterdam, Netherlands
- 2Obstetrics, Chelsea and Westminster Hosp, London, United Kingdom
- 3Cardiology, Norwich Med Sch, Univ of East Anglia, Norwich, United Kingdom
Abstract 18498: Description of Aortic Root Growth and Outcomes in a Cohort of Pediatric Patients with Loeys-Dietz Syndrome
Nitya Viswanathan, Shaine A Morris
Circulation. 2014;130:A18498
Abstract
Background: Loeys Dietz Syndrome (LDS) is associated with rapid aortic dilation and aortic dissection, but data on children with LDS are limited. The goal of this study is to describe aortic root growth and outcomes in children with LDS.
Methods: Patients with LDS were identified from an institutional database. Data regarding genetic mutation, medications, aortic root dimensions by transthoracic echocardiography (TTE), aortic dissection and surgical intervention were collected. For those with >2 TTEs 1 year apart, rate of change in z-score was calculated using linear regression. TTEs performed after aortic surgery were excluded. We examined if variables were associated with rate of aortic root growth.
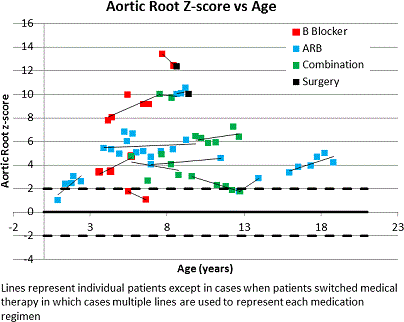
Results: Of 16 patients, 8 were female. Five had a TGFBR1 and 9 had a TGFBR2 mutation; 2 patients did not have genetic data available. Median aortic root Z-score at diagnosis was 3.5 (range 0.5-25.4). Fifteen patients were on medication (2 beta-blocker (BB), 5 angiotensin receptor blocker (ARB), 5 BB+ARB, and 3 with past use of both). Four patients underwent prophylactic root replacement at ages 3.3, 6.7, 8.7, and 9.4 years at root dimensions 3.2, 6.5, 4.0, and 4.1 cm respectively. One patient had a Type A dissection at age 15y after prior root replacement and underwent repeat surgery. Another underwent heart transplant at 6.9 years old after prior root replacement. Ten patients had serial TTE data. Median change in aortic root diameter and Z-score was 0.11cm/year and 0.1/year respectively. Mean change in z-score per year for those on on BB was -0.1± (range -1.2 to 0.7), ARB 0.5 (range 0.1 to 1.1) and both 0.0 (range -0.2 to 0.2, p=NS). No variables studied were associated with faster aortic growth.
Conclusions: Degree of aortic root dilation and rate of aortic root growth is highly variable in children with LDS, although factors associated more aggressive disease are unclear. The high proportion of patients with adverse outcomes including aortic dissection and surgery is concerning.
Article Information
vol. 130 no. Suppl 2 A18498
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Nitya Viswanathan;
- Shaine A Morris
- Pediatrics, Texas Childrens Hosp, Houston, TX
Abstract 18062: Neonatal Outcomes and Maternal Morbidity Associated With Pregnancy in Women with Congenital Heart Disease
Camden Hebson, Dan Lin, Turgay Ayer, Jan Vlachy, Martina Badell, Marissa Platner, Agasha Katabarwa, Julia Shinnick, Anurag Sahu, Wendy Book
Circulation. 2014;130:A18062
Abstract
Background: Women with congenital heart disease (WCHD) who are considering pregnancy need counseling regarding maternal and offspring risk. Previous studies have not focused on neonatal outcomes and have mostly described lower risk patients.
Methods and Results: We enrolled 113 WCHD (mean age 32 ± 5 years, 51% with NYHA functional class >1, 28% with World Health Organization (WHO) risk class > 2, 81% with moderate or higher CHD complexity) in our study and reviewed the medical record for their baseline cardiac status as well as their post-natal outcomes. We compared our cohort to a demographically similar cohort of patients (n = 147) without CHD. Neonatal outcomes included prematurity (< 37 weeks gestation), admission to the NICU, low birth weight (< 2.5 kg), and neonatal death, while maternal outcomes were arrhythmias, heart failure, stroke, and death. Significant predictors of neonatal complications included history of cyanotic CHD (OR 3.1, CI 1.0-9.7, p = 0.04), need for cardiac medications (OR 3.2, CI 1.0-10.2, p = 0.04), and WHO class > 2 (OR 3.5, CI 1.1-11.1, p = 0.03). WHO class (OR 4.0, CI 1.1-15.2, p = 0.03) independently predicated maternal complications. Compared to controls, neonates born to WCHD were more likely to be premature (29 vs. 7%, p < 0.01), low birth weight (27 vs. 3%, p < 0.01), and admitted to the NICU (19 vs. 8%, p = 0.01).
Conclusions: Preconception counseling for women with CHD should include assessment of neonatal risk in addition to maternal risk. While women with more complex CHD can complete pregnancy successfully, both mother and offspring must endure higher odds of complications.
Article Information
vol. 130 no. Suppl 2 A18062
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Camden Hebson1;
- Dan Lin2;
- Turgay Ayer3;
- Jan Vlachy3;
- Martina Badell4;
- Marissa Platner4;
- Agasha Katabarwa5;
- Julia Shinnick1;
- Anurag Sahu1;
- Wendy Book1
- 1Medicine, Emory Univ Sch of Medicine, Atlanta, GA
- 2Rollins Sch of Publich Health, Emory Univ Sch of Medicine, Atlanta, GA
- 3H. Milton Stewart Sch of Industrial and Systems Engineering, Georgia Institute of Technology, Atlanta, GA
- 4Gynecology and Obstetrics, Emory Univ Sch of Medicine, Atlanta, GA
- 5Rollins Sch of Public Health, Emory Univ Sch of Medicine, Atlanta, GA
Abstract 11618: Low Antithrombin Level Correlates With Failing Fontan Circulation and Predicts the Mortality
Nobuyuki Tsujii, Hideo Ohuchi, Yosuke Hayama, Jun Negishi, Kanae Noritake, Osamu Sasaki, Aya Miyazaki, Osamu Yamada
Circulation. 2014;130:A11618
Abstract
Background: Antithrombin is one of natural anticoagulants produced by the liver and the activity (AT) decreases as a result of liver dysfunction. Fontan pathophysiology includes heart failure-related hepatopathy that accompanies coagulation abnormality.
Purpose: To clarify clinical significance of AT in Fontan patients.
Methods and Results: We prospectively measured AT in consecutive 303 Fontan patients (17±9 years) and compared with the clinical variables, including hemodynamics, peak oxygen uptake (PVO2), liver function, plasma brain natriuretic peptide (BNP), and unscheduled hospitalization (USH), including all-cause mortality. The AT was 109±14 (%, normal range: 80-120%) and it correlated inversely with age, central venous pressure (CVP), and plasma levels of BNP and creatinine, and positively with arterial oxygen saturation (Sat) and PVO2 (p < 0.001 for all). Patient with heterotaxy syndrome (HS) showed a low AT (p < 0.001). The lower plasma albumin level and platelet count and high total bilirubin level were associated with the lower AT (p < 0.05-0.0001). Multivariate analysis revealed that HS, older age, high levels of CVP, total bilirubin and BNP, and low Sat independently associated with the low AT. During the follow-up of 26 months, 53 USH, including 14 deaths, occurred. Low AT predicted USH (HR: 0.72 per 10 %, 95%CI: 0.60-0.88, p = 0.0012), especially the mortality (HR: 0.40 per 10 %, 95%CI: 0.28-0.56, p < 0.0001).
Conclusions: Low AT was an ominous clinical manifestation that closely associated with failing Fontan circulation in adults, especially those with HS, and predicted the morbidity and morbidity.
Article Information
vol. 130 no. Suppl 2 A11618
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Nobuyuki Tsujii;
- Hideo Ohuchi;
- Yosuke Hayama;
- Jun Negishi;
- Kanae Noritake;
- Osamu Sasaki;
- Aya Miyazaki;
- Osamu Yamada
- Pediatric Cardiology, National Cerebral and Cardiovascular Cntr, Osaka, Japan
Abstract 19493: Hybrid Alternatives to Norwood Stage-1 Are Not a Lower Risk Alternative: Norwood-RVPA Offers Better Outcome in Comparable Neonates
Travis J Wilder, Edward J Hickey, Gerhard Ziemer, Christo I Tchervenkov, Marshall L Jacobs, Peter J Gruber, Eugene H Blackstone, Brian W McCrindle, William G Williams, William M DeCampli, Christopher A Caldarone, Christian Pizarro
Circulation. 2014;130:A19493
Abstract
Introduction: Hybrid strategies (HYBRID) for critical LVOTO have emerged as alternatives to stage-1 Norwood-BT shunt (BT) or Norwood-RV to PA conduit (RVPA) and may be pursued in neonates with unfavorable risk profile. An RCT determining the merit of HYBRID seems unlikely. We investigated the potential survival advantage of HYBRID strategies.
Methods: In an inception cohort of neonates with critical LVOTO (2005-2014; 21 institutions) 564 had initial surgical palliation consisting of; stage-1 HYBRID (110; 20%), BT (232; 41%) or RVPA (222; 39%). Risk of death without Fontan/transplant was analyzed using risk-adjusted parametric competing risks models. Additional comparisons between HYBRID and BT/RVPA were made via propensity-matching using baseline morphologic and demographic variables.
Results: At 6-years post stage-1, 49% and 8% had transitioned to Fontan and transplant respectively, 9% were alive without transition and 34% died; mortality plateaued at 3-years. Risk factors for death included small/atretic LVOT, small branch pulmonary arteries and low birth weight (BW). HYBRID was associated with a lower median BW (kg) than BT or RVPA (p<.01). RVPA was a strong predictor of decreased death (22%, p<.01) versus HYBRID or BT (both 36%; figure).
Matched Comparison:
Propensity matching resulted in 82 paired HYBRID/BT neonates (c-statistic=.77). Risk-adjusted 3-year mortality tended to be lower for HYBRID (32% vs. 40%, P=.28). Matching between HYBRID and RVPA resulted in 82 pairs (c-statistic=.75). Risk-adjusted mortality was significantly higher for HYBRID (42% vs. 25%; p=0.02). However, at low BW HYBRID had a survival advantage over RVPA at <~2kg and BT at <~3kg (figure).
Conclusions: In neonates with critical LVOTO RVPA offers better survival to Fontan. Although baseline characteristics suggest an appropriate bias towards HYBRID in some higher risk neonates (e.g. low BW), HYBRID may not be lower risk alternative to Norwood in otherwise equivalent patients.
Article Information
vol. 130 no. Suppl 2 A19493
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Travis J Wilder1;
- Edward J Hickey2;
- Gerhard Ziemer3;
- Christo I Tchervenkov4;
- Marshall L Jacobs5;
- Peter J Gruber6;
- Eugene H Blackstone7;
- Brian W McCrindle8;
- William G Williams1;
- William M DeCampli9;
- Christopher A Caldarone2;
- Christian Pizarro10
- 1CHSS Data Cntr, The Hosp For Sick Children, Toronto, Canada
- 2Div of Cardiovascular Surgery, The Hosp For Sick Children, Toronto, Canada
- 3Pediatric Cardiac Surgery, Univ of Chicago Med Cntr, Chicago, IL
- 4Div of Cardiovascular Surgery, Montreal Children’s Hosp of the McGill Univ Health Cntr, Montreal, Canada
- 5Div of Cardiac Surgery, Johns Hopkins, Newtown Square, PA
- 6Dept of Cardiothoracic Surgery, Univ of Iowa Carver College of Medicine, Iowa City, IA
- 7Dept of Thoracic and Cardiovascular Surgery, Clevleland Clinic, Cleveland, OH
- 8Dept of Cardiology, The Hosp For Sick Children, Toronto, Canada
- 9Pediatric Cardiovascular Surgery, The Heart Cntr at Arnold Palmer Hosp for Children and the Univ of Central Florida College of Medicine, Orlando, FL
- 10Dept of Cardiovascular Surgery, Nemours Cardiac Cntr. Alfred I. duPont Hosp for Children, Wilmington, DE
Abstract 13233: Does Fetal Aortic Valvuloplasty alter the Natural History of Aortic Stenosis?
Alexander Kovacevic, Mats Mellander, Gerald Tulzer, Ulrike Herberg, Joanna Dangel, Annika Öhman, Helena Gardiner
Circulation. 2014;130:A13233
Abstract
Introduction: Fetal aortic valvuloplasty (FV) is proposed as therapy to achieve biventricular circulation (BV) in fetuses where univentricular circulation (UV) is likely.
Hypothesis: FV cannot alter natural history (NH) outcome.
Methods: Hybrid of case-control and repeated samples cohort study. Fetuses with aortic stenosis (AS) were enrolled in a multicenter study (2005-2012). FV was considered in 70 / 214 AS and successful in 59/67 (88.0%) performed. Six salvage cases (hydrops) were excluded and 47 liveborn FV could be matched with 95 controls (NH) by scan closest to 23 +/- 3 weeks and +/- 1 Z-score for MV, LV and AV, producing a best match group for each.
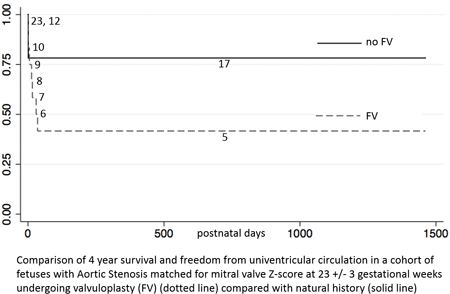
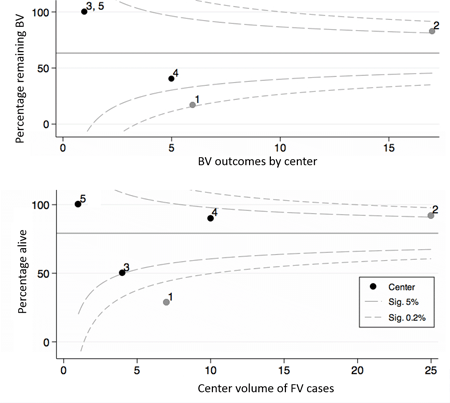
Results: Procedure-related death occurred in 7/67 (10.4%). Overall 151/214 (71%) were liveborn, but outcome unknown in 5. Serial left sided growth was similar in FV and NH: Z score differences MV = 0.11, LV = 0.08, AV = 0.11, p>0.90. Hazard ratio for FV survival was similar to NH at 30 d, 1 and 4 yrs after birth [0.68 (95% CI 0.347 – 1.315), p= 0.25]. Cohorts matched for MV, LV and AV did not show survival advantage after FV and survival with freedom from UV circulation showed fewer BV survivors in FV than NH. (Fig 1) Funnel plots show improved BV survival by center volume for FV, but more BV-UV conversions in one with limited surgical options where 17% vs 82% FV remain BV. (Fig 2)
Conclusions: Data show no survival advantage or improved chance of BV at 4 years in fetuses matched for morphology at 23 wks undergoing FV. Centralization of FV may improve survival, but BV – UV conversion suggests a specialized surgical approach is also essential to maintain BV outcome. A carefully designed prospective study is indicated to better evaluate FV. procedure.
Article Information
vol. 130 no. Suppl 2 A13233
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Alexander Kovacevic1;
- Mats Mellander2;
- Gerald Tulzer3;
- Ulrike Herberg4;
- Joanna Dangel5;
- Annika Öhman6;
- Helena Gardiner7
- 1Cntr for Fetal Care, Imperial College London, London, United Kingdom
- 2Dept of Paediatrics, Queen Silva Children’s Hosp, Gothenburg, Sweden
- 3Dept of Pediatric Cardiology, Childrens’ Heart Cntr Linz, Linz, Austria
- 4Dept of Paediatric Cardiology, Children’s Hosp, Univ Hosp of Bonn, Bonn, Germany
- 5Perinatal Cardiology, Med Univ of Warsaw, Warsaw, Poland
- 6Dept of Paediatric Cardiology, Astrid Lindgren Children’s Hosp, Stockholm, Sweden
- 7Texas Fetal Cntr, Univ of Texas at Houston, Houston, TX
Abstract 11789: Intracoronary Delivery of Autologous Cardiac Progenitor Cells in Children With Hypoplastic Left Heart Syndrome: The Ticap Trial With 18-Month Follow Up
Shuta Ishigami, Suguru Tarui, Michihiro Okuyama, Daiki Ousaka, Shinichi Ohtsuki, Takahiro Eitoku, Junko Kobayashi, Shingo Kasahara, Shunji Sano, Hidemasa Oh
Circulation. 2014;130:A11789
Abstract
Backgrounds: Hypoplastic left heart syndrome (HLHS) is one of the severe malformations in congenital heart disease. This study is to investigate whether intracoronary delivery of autologous cardiosphere-derived cells (CDCs) is feasible and safe to treat the children with HLHS.
Methods and Design: This phase 1 trial (TICAP: NCT01273857) is a prospective controlled study. Four-teen patients with HLHS who are undergoing staged-2 or -3 surgical palliations were enrolled between January, 2011, and January, 2012. Seven patients assigned to receive intracoronary CDCs infusion 1 month after the shunt procedures followed by 7 patients allocated to a control group with standard palliations. The primary endpoint was to assess the safety and the secondary endpoint was to evaluate the cardiac function and heart failure status from the baseline through 18 months of follow-up.
Results: No complications, including cardiac death, myocardial ischemia, arrhythmia, re-hospitalization, and tumor formation, were reported within 18 months after CDCs infusion. Echocardiography showed that the absolute improvement of right ventricular ejection fraction (RVEF) was greater in the CDCs-treated group (+7.1±5.5%) than in controls (+2.1±0.7%, P=0.04) at 18 months. Compared with controls, cMRI analysis showed that patients with CDCs infusion had significantly increased RVEF (31.5±6.8% vs 40.4±7.6%, P=0.04) and reduced end-systolic volume index at 18 months (P=0.049). The improved mechanical output was addressed by a gain of end-systolic elastance (P=0.03) and ventriculo-arterial coupling (P=0.02) in CDC-treated group at 18-month compared with baseline. The increased cardiac performance in long-term resulted in greater somatic growth (weight-for-age z score, P=0.02), reduced heart failure status assessed by Ross classification (P=0.004) and NYU PHF index (P=0.04), decrease in BNP levels (P=0.049), and significantly lower incidence of coil occlusion for collaterals (P=0.007) by 18 months after CDC transfer than controls.
Conclusion: Transcoronary infusion of CDCs appeared to be feasible and safe to treat the patients with HLHS. These initial results provide the basis for larger studies to assess the efficacy of this novel therapeutic approach in children.
Article Information
vol. 130 no. Suppl 2 A11789
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Shuta Ishigami1;
- Suguru Tarui1;
- Michihiro Okuyama1;
- Daiki Ousaka1;
- Shinichi Ohtsuki2;
- Takahiro Eitoku2;
- Junko Kobayashi1;
- Shingo Kasahara1;
- Shunji Sano1;
- Hidemasa Oh3
- 1Dept of Cardiovascular surgery, Okayama university hospital, Okayama, Japan
- 2Dept of Pediatrics, Okayama university hospital, Okayama, Japan
- 3Dept of of Regenerative Medicine, Cntr for Innovative Clinical Medicine, Okayama university hospital, Okayama, Japan
Abstract 19933: Late Fontan Completion Associates with a Reduced Distensibility of the Ascending Aorta in Long-term Fontan Survivors
Yohsuke Hayama, Hideo Ohuchi, Aya Miyazaki, Satoshi Yazaki, Etsuko Tsuda, Osamu Yamada
Circulation. 2014;130:A19933
Abstract
Background: Stiffened and dilated ascending aorta (AAo) before and long after operation, which may be an important predictor of cardiovascular morbidity and mortality has been reported in Fontan patients. However, the determinant of reduced distensibility has not been clarified.
Methods: Ninety nine postoperative F patients followed-up over 15 years were included (age at Fontan ; 4.5 +/- 5.2 years old). All patients underwent cardiac catheterization with aortography before and 15 years after operation (F0, F15). We measured the diameters of the AAo and descending aorta (DAo) from the cine-angiogram, and standardized by body height. We also calculated the stiffness parameters of the AAo and DAo (β(AAo),β(DAo)) with the corresponding systolic and diastolic pressures and distensible change of diameters. We compared the β(AAo) at F15 with clinical variables, including hemodynamics before and 15-year after the F operation.
Results: In the univariate analysis, β(AAo) at F15 was positively correlated with age at F operation (p < 0.001), β(DAo) at F15 (p = 0.025), β(AAo) at F0 (p = 0.026) , end-diastolic pressure of systemic ventricle (EDP) at F15 (p = 0.019), number of surgical interventions before F operation (p < 0.001), and was negatively correlated with ejection fractions at F0 (p = 0.047) and F15 (p = 0.047). In the multivariate analysis, age at F operation (p = 0.004), number of surgical interventions before F operation (p < 0.001), EDP at F15 (p = 0.036) were independently correlated with β(AAo) at F15.
Conclusions: Delayed Fontan completion leads to the stiffened ascending aorta and may result in increase in end-diastolic pressure of ventricle long after operation. Early Fontan completion may prevent progressions of stiffened aorta and ventricular dysfunction.
Article Information
vol. 130 no. Suppl 2 A19933
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Yohsuke Hayama;
- Hideo Ohuchi;
- Aya Miyazaki;
- Satoshi Yazaki;
- Etsuko Tsuda;
- Osamu Yamada
- Pediatric Cardiology, National Cerebral and Cardiovascular Cntr, Suita, Japan
Abstract 12308: Assessment of Liver Fibrosis Using Magnetic Resonance Elastography in Children Who Undergo the Fontan Procedure and Intracardiac Repair
Masaya Sugimoto, Hideharu Oka, Aya Kajihama, Kouichi Nakau, Hiroshi Azuma
Circulation. 2014;130:A12308
Abstract
Background: The incidence of late complications related to the liver such as fibrosis/cirrhosis is increasing in patients who have undergone the Fontan procedure and may contribute to morbidity and mortality. Recently, magnetic resonance elastography (MRE), a novel evaluation technique of liver fibrosis, has been attracting attention. However, few reports have described the use of MRE for evaluating liver fibrosis in children with congenital heart disease (CHD).
Methods: Thirty-two children were examined and divided into 4 groups: 12 children with CHD who underwent intracardiac repair (ICR; median age, 13.0 years); 10 with CHD who underwent the Fontan procedure (Fontan; 15.3 years); 8 who were included in the control group (control; 15.8 years); and 2 children with cirrhosis (cirrhosis; 16.3 years). The liver stiffness (LS) was estimated by MRE. LS was measured 3 times consecutively, and the mean value was considered for further analysis. Central venous pressure (CVP) and cardiac index (C.I.) were determined by cardiac catheterization. The levels of cardiac biomarkers (NTproBNP and PIIIP) were determined at the same time.
Results: Among the 4 groups, no significant differences were observed in age, C.I., and NTproBNP levels. The PIIIP levels in the cirrhosis group were significantly higher than those in the control, but no significant difference in PIIIP levels was found among the other groups (p < 0.01). LS in the Fontan and cirrhosis group was significantly higher than that in the control group (5.6, 15.3 vs. 2.4 kPa, respectively; Fig. 1). Furthermore, there was a strong correlation between LS and CVP (r = 0.802; Fig. 2).
Conclusions: This study showed that LS is a direct function of CVP, which should be considered when assessing the degree of liver fibrosis in children with CHD. In particular, in the case of children who undergo the Fontan procedure, the highly sensitive MRE can be used to evaluate liver fibrosis and help detect LS earlier than cardiac biomarkers do.
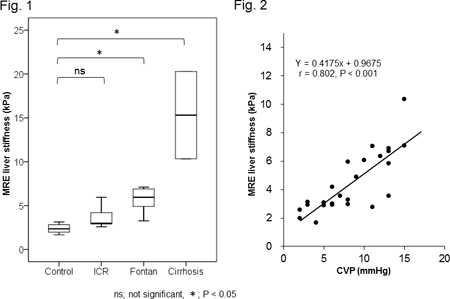
Article Information
vol. 130 no. Suppl 2 A12308
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Masaya Sugimoto;
- Hideharu Oka;
- Aya Kajihama;
- Kouichi Nakau;
- Hiroshi Azuma
- Pediatrics, Asahikawa Med Univ, Asahikawa, Japan
Abstract 12145: Cardiac Magnetic Resonance Late Gadolinium Enhancement is Associated With Ventricular Elastance That May Predict Latent Ventricular Dysfunction After Fontan Procedure
Michihiro Okuyama, Shuta Ishigami, Daiki Ousaka, Junko Kobayashi, Sadahiko Arai, Shingo Kasahara, Shunji Sano, Hidemasa Oh
Circulation. 2014;130:A12145
Abstract
Backgrounds: Systemic right ventricular circulation after Fontan procedures is known to have late hemodynamic complications. Although a number of studies have investigated the factors that may impact on survival, postoperative outcomes after palliations remain to be elucidated.
Objective: The purpose of this study is to investigate the prognostic value of myocardial fibrosis identified by cardiac magnetic resonance imaging (cMRI) in patients with single ventricular physiology.
Methods: Consecutive 23 patients undergoing Fontan procedures were prospectively scheduled to have cMRI study with late gadolinium enhancement (LGE) imaging and ventricle circumferential strain measurement before and 4 months after Fontan operation.
Results: Of 23 patients (mean age 3.3±0.9 years), 7 were positive for LGE (LGE+) and median percent LGE was 3.0% (interquartile range 3.0% to 7.5%). Pre-Fontan examinations revealed that patients with LGE+ showed an increase in end-diastolic volume index (139.7±26.8 ml/BSA vs. 113.3±20.9 ml/BSA; P=0.02) and end-systolic volume index (ESVI: 99.9±32.2 ml/BSA vs. 70.8±20.0 ml/BSA; P=0.01) compared with those without LGE (LGE-). In contrast to LGE- group, LGE+ patients showed lower global circumferential strain (4.1±2.3% vs. 7.9±2.7%, P=0.006), decreased ejection fraction (EF: 29±9.1% vs. 38±8.7%; P=0.04), and reduced end-systolic elastance (1.1±0.3 mm Hg/ml/m2 vs. 1.7±0.5 mm Hg/ml/m2). In addition, LGE+ group had higher levels of BNP (91.0±72.4 pg/ml vs. 30.9±44.0 pg/ml, P=0.02) and New York University Pediatric Heart Failure Index (10.9±3.3 vs. 7.8±1.1, P=0.02) than LGE- group. This was validated by positive correlations between the area of LGE versus ESVI (r=0.85, P=0.01) and BNP levels (r=0.82, P=0.02), respectively. At 4 months after Fontan procedure, LGE- group showed higher EF (37.5±8.6% vs. 24.0±8.9%, P=0.02) compared with those in LGE+ patients, and increased global circumferential strain (6.5±2.0% to 7.4±2.7%, P=0.04).
Conclusion: LGE identified by cMRI before operation may be associated with lower ventricular elastance that resulted in poorer functional recovery after staged palliation. This novel strategy may provide a prognostic value of latent myocardial dysfunction after Fontan procedure.
Article Information
vol. 130 no. Suppl 2 A12145
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Michihiro Okuyama1;
- Shuta Ishigami1;
- Daiki Ousaka1;
- Junko Kobayashi1;
- Sadahiko Arai1;
- Shingo Kasahara1;
- Shunji Sano1;
- Hidemasa Oh2
- 1Cardiovascular Surgery, Okayama Univ Hosp, Okayama, Japan
- 2Regenerative Medicine, Cntr for Innovative Clinical Medicine, Okayama Univ Hosp, Okayama, Japan
Abstract 11430: Clinical Significance of Impaired Vascular Endothelial Function in Patients After Fontan Procedure
Seiji Asagai, Kei Inai, Hirofumi Tomimatsu, Tokuko Shinohara, Toshio Nakanishi
Circulation. 2014;130:A11430
Abstract
Background: It has been reported that patients have reduced vascular endothelial function after Fontan procedure; however, its underlying mechanisms and correlation with exercise capacity are not widely known.
Purpose: We evaluated vascular endothelial function (flow-mediated dilatation [FMD]) in patients after Fontan procedure using conventional vascular ultrasonography and investigated the correlation between FMD and clinical profile.
Patients and Methods: Thirty-one patients who underwent the Fontan procedure (age, 12-55 years; median age, 20 years; postoperative duration, 9.5-31.8 years; median postoperative duration, 16.9 years) and 17 age- and sex-matched healthy subjects were enrolled in this study. We measured FMD in these patients. We examined the correlations between FMD and age, duration after the Fontan procedure, laboratory results (total cholesterol, triglyceride, hemoglobin A1c, fibrinogen and brain natriuretic peptide levels), hemodynamic profiles (central venous pressure, end-diastolic pressure, cardiac index, pulmonary vascular resistance, arterial oxygen saturation, systolic blood pressure), arterial stiffness index (pulse wave velocity [PWV], ankle brachial pressure index [ABI], stiffness parameter [β], elastic modulus [Ep]), and exercise tolerance (6-min walk distance).
Results: Patients who underwent the Fontan procedure had a lower FMD than healthy subjects (7.8 ± 1.9% vs. 11.0 ± 1.7%; p < 0.001). FMD showed a negative correlation with age, duration after the Fontan procedure (r, -0.68, p < 0.001 and r, -0.44, p = 0.014 respectively) and total cholesterol (r, -0.44, p = 0.020), but no significant correlation with hemodynamic characteristics. Considering arterial stiffness index, FMD showed a negative correlation with β and Ep (r, -0.65, p < 0.001 and r, -0.65, p < 0.001, respectively), but no significant correlation with PWV or ABI. FMD showed a positive correlation with 6-min walk distance (r, 0.38, p = 0.039).
Conclusions: Vascular endothelial dysfunction after the Fontan procedure contributes to reduced exercise tolerance and is likely related to collapse of the Fontan circulation. Vascular endothelial dysfunction is due to both a combination of Fontan hemodynamics and metabolic disorders.
Article Information
vol. 130 no. Suppl 2 A11430
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Seiji Asagai;
- Kei Inai;
- Hirofumi Tomimatsu;
- Tokuko Shinohara;
- Toshio Nakanishi
- Pediatric Cardiology, Tokyo Women’s Med Univercity, Tokyo, Japan
Abstract 11089: Hepatic Abnormalities are Present Before and Early After the Fontan Operation
Matthew Schwartz, Andrew C Glatz, Kaitlyn Daniels, David J Goldberg, Elizabeth Rand, Monica Epelman, Meryl S Cohen
Circulation. 2014;130:A11089
Abstract
Objectives: Progressive hepatic fibrosis is common after the Fontan operation, but little is known about its onset. We sought to determine if there is non-invasive evidence of hepatic injury prior to the Fontan operation, and if further injury is seen soon after the procedure.
Methods: Patients undergoing the Fontan operation at our institution were prospectively enrolled and underwent hepatic ultrasound with Doppler and serum testing immediately before and 3 to 6 months after the operation.
Results: Thirty patients were enrolled at a median age at time of the Fontan operation of 3.1 yrs (range: 2.2-8.1 yrs). An extracardiac Fontan operation was performed in 67% and nearly all (97%) underwent fenestration. Three patients (10%) had abnormal hepatic echotexture prior to the Fontan operation. At the post-Fontan study, mean liver length increased (9.9 vs. 10.9 cm, p<0.0001) and mean hepatic artery end diastolic velocity decreased (18.8 vs. 14.5 cm/sec, p=0.03). One patient showed new, abnormal hepatic echotexture after surgery. Among serum indices, mean aspartate aminotransferase (56.7 vs. 60.7 IU, p=0.04), mean alanine transaminase (ALT) (18.9 vs. 33.9 IU, p=0.0002), and mean gamma-glutamyl transferase (GGT) (18.7 vs. 46.1 IU, p=0.002) increased at the post-Fontan assessment compared to the pre-operative values. By linear regression, hospital length of stay and duration with chest tube after Fontan operation were both significantly associated with an increase in GGT (p< 0.001 for both) and ALT (p=0.008, p=0.04) 3 to 6 months after surgery. There were no associations found between change in ultrasound or serum markers of liver function and pre-Fontan hemodynamic variables as measured by echocardiogram, catheterization, and/or magnetic resonance imaging.
Conclusions: Hepatic ultrasound abnormalities were seen prior to the Fontan operation in some patients. Early after the Fontan operation, liver length and serum hepatic markers were increased relative to pre-Fontan values. Post-operative morbidity was associated with an increase in these serum markers. In total, these findings suggest that liver insult may occur prior to the Fontan operation and further insult likely begins soon after the Fontan circulation is created.
Article Information
vol. 130 no. Suppl 2 A11089
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Matthew Schwartz1;
- Andrew C Glatz1;
- Kaitlyn Daniels1;
- David J Goldberg1;
- Elizabeth Rand2;
- Monica Epelman3;
- Meryl S Cohen1
- 1PEDIATRIC CARDIOLOGY, Childrens Hosp Philadelphia, Philadelphia, PA
- 2PEDIATRIC Gastroenterology, Childrens Hosp Philadelphia, Philadelphia, PA
- 3Pediatric Radiology, Nemours Children’s Hosp, Orlando, FL
Abstract 20578: Absence of Family History Predicts a Low Yield for Genetic Testing in a Community Cardiomyopathy Clinic
Nikhil Mehta, Mark Marieb, Lavanya Bellumkonda, Daniel Jacoby
Circulation. 2014;130:A20578
Abstract
Objective: Although guidelines exist suggesting indications for genetic testing in inherited cardiomyopathy, the yield of testing outside specialized quarternary referral centers remains unexplored. We aimed to assess the yield of genetic testing in a community cardiomyopathy clinic. We further aimed to assess the impact of presence or absence of family history (broadly defined) on the yield of genetic testing.
Methods: Clinical characteristics and genetic results for all patients seen in the Yale Cardiomyopathy Clinic were prospectively entered into a clinical database (n=771) over 5 years. 209 (27.1%) underwent genetic testing to determine the presence of pathogenic mutations. 43 family screens were excluded from analysis yielding a total n of 166. Pathogenic mutations were defined by the destination lab at the time of reporting. For the purposes of this analysis family history was defined as presence of any of the following on 3 generation pedigree: cardiomyopathy, pacemaker, defibrillator, sudden cardiac death, LVAD and/or heart transplant.
Results: Of those undergoing genetic testing, 71 (42.8%) had hypertrophic cardiomyopathy (HCM), 51 (30.7%) had dilated cardiomyopathy (DCM), 9 (5.4%) had arrhythmogenic ventricular cardiomyopathy (ARVC/ALVC) and 35 (21.1%) had unclassified cardiomyopathy. Pathogenic mutations were found in 38% of HCM cases (27/71), 35.3% of DCM cases (18/51), 33.3% of ARVC/ALVC cases (3/9) and 20% of unclassified (7/35). A negative family history predicted a poor yield (9/57, 15.8%) of pathogenic mutations versus a positive family history (46/109, 42.2%) with a p value of 0.0005.
Conclusion: Absence of family history predicts a low yield for genetic testing in a community cardiomyopathy clinic. As genetic testing moves into community practice, presence or absence of broadly defined family history may guide physicians in counseling their patients about the yield of potentially costly, complex testing.
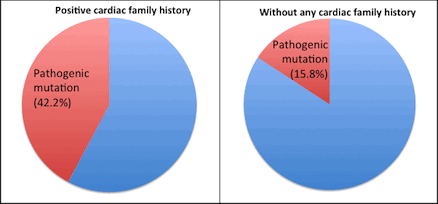
Article Information
vol. 130 no. Suppl 2 A20578
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Nikhil Mehta;
- Mark Marieb;
- Lavanya Bellumkonda;
- Daniel Jacoby
- Cardiovascular, Yale Sch of Medicine, New Haven, CT
Abstract 20426: Study of S-Nitrosylation in Duchenne Muscular Dystrophy
Heaseung S Chung, Peter P Rainer, David A Kass, Jennifer E Van Eyk
Circulation. 2014;130:A20426
Abstract
Introduction: Severe cardiac myopathy occurs with Duchenne muscular dystrophy (DMD), a fatal disease characterized by absence of functional dystrophin protein. The mechanisms of the disease in cardiac muscle and whether it mimics some of the pathophysiological mechanisms present in skeletal muscle are not clear. The loss of dystrophin was reported to disrupt neuronal nitric oxide synthase mu, presumably altering redox-signaling. Nitric oxide-induced S-nitrosylation (SNO) on cysteine residues is known as a redox-sensor, which responds to cellular dynamic redox-environment. Here we identified and quantified the SNO-modified sites of cardiac tissue in dystrophy model using a novel proteomics approach.
Methods: SNO-proteins from cardiac tissues of 10 months-old mdx/utrn+/- (n=3) and C57BL/6 (n=3) mice were detected with our improved TMT (tandem mass tag)-switch technique coupled with liquid chromatography/tandem mass spectrometry. The identified SNO of each site was quantified across DMD and WT samples and the fold change of SNO on each site in DMD compared to WT was displayed as ‘SNO-index (I)’, calculated by SNODMD/SNOWT.
Results: Proteomics analysis identified total 866 SNO-modified peptides on 287 SNO-proteins in the dystrophy cardiac tissue. The magnitudes of SNO-fold change were various across peptides (I=0.1 least to 1702.7 most). 32% of these, 278 peptides were more than two-fold oxidized in DMD than WT. Interestingly, a termed ‘Dilated cardiomyopathy’ protein group by KEGG-pathway analysis has extremely oxidized sites than other groups, which are more than 10-folds S-nitrosylated in DMD. Myosin-6 at Cys949 and Cys1750 (I=11.4, 117.3), Cys432 of myosin-binding protein C (I=135.0) and Cys191 of myosin light chain 3 (I=81.2) displayed a considerable SNO-increase in dystrophy sample and Cys35 of troponin C was also more modified (I=2.9), while other sites such as Cys397 of creatine kinase S-type was less oxidized (I=0.26) in DMD.
Conclusions: Our novel TMT-switch assay allowed quantification of the very labile SNO-modification as well as site-identification. More than 500 cysteine residues sensed the oxidative stimuli and each had a specific susceptibility to SNO. The SNO-based redox switches were shifted in DMD compared with WT.
Article Information
vol. 130 no. Suppl 2 A20426
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Biological Chemistry, Johns Hopkins Univ, Baltimore, MD
- 2Cardiology, Johns Hopkins Univ, Baltimore, MD
Abstract 19609: Novel Wnt Signaling in Neonatal Heart During Perinatal Transition
Marlin Touma, Xuedong Kang, Ashly Cass, Xinshu Xiao, Yibin Wang
Circulation. 2014;130:A19609
Abstract
Background: Fetal to neonatal transition of the mammalian heart is an elaborate process, during which the neonatal cardiomyocytes undergo complete maturation, differentiation, and irreversible exit from the cell cycle. However, the molecular mechanisms that control the chamber specific- growth of postnatal heart are understudied. In particular, the permanent arrest of cardiomyocytes cycling remains mysterious both in terms of the etiology and the mechanisms.
Objectives: To determine factors of chamber specificity in neonatal heart during postnatal growth and maturation.
Methods: Deep RNA-seq (2x92nt) was performed on male newborn mouse left ventricle (LV) and right ventricle (RV) at P0, P3 and P7. Neonatal Rat Ventricular Myoctes (NRVM), Wnt11-siRNA, and recombinant Wnt11 (rWnt11) were used to achieve Wnt11 Inhibition and gain of function respectively In Vitro. Wnt11-Antisense oligo nucleotide was used to achieve In Vivo inhibition of Wnt11. Anti-Phospho histone3 (Ph3) was used to assess proliferation.
Results: Extensive transcriptome analysis and experimental validation revealed distinct, chamber specific- patterns of temporal regulation of cell cycle genes and Wnt signaling during maturation. Specifically, an RV specific- induction of cell cycle genes was found to reciprocally correlate with a robust LV specific- enrichment of the non-canonical-Wnt11 at P7. Functional studies revealed enhancement of NRVM maturation markers and down-regulation of cell cycle genes in response to Wnt11 gain of function. In contrast, Wnt11 inhibition induced NRVM proliferation markers and reduced the size and the fraction of bi-nucleated cells supporting a functional impact of wnt11. Furthermore, InVivo inhibition of Wnt11 at late gestation enhanced Ph3 and cell cycle markers at P0 suggesting cellular proliferation.
Conclusions: Our findings suggest novel molecular basis for chamber-specific programming of proliferation and maturation in neonatal heart, including differential enrichment of cell cycle genes and novel Wnt11-mediated signaling. Further mechanistic studies to decipher putative roles of Wnt11 signaling in LV vs. RV programming during maturation in Vivo will likely pave the way to novel chamber-specific therapeutic targets.
Article Information
vol. 130 no. Suppl 2 A19609
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Marlin Touma;
- Xuedong Kang;
- Ashly Cass;
- Xinshu Xiao;
- Yibin Wang
- Pediatrics, Cardiovascular Rsch Laboratory, Univ of Clifornia Los Angeles, David Geffen Sch of Medicine, Los Angeles, CA
Abstract 18637: Differing Genetic Expression Patterns in Infants with Aortic Coarctation With Without Arch Hypoplasia
Michael F Swartz, Pratik Parikh, Jill M Cholette, Nader Atallah-Yunes, George M Alfieris
Circulation. 2014;130:A18637
Abstract
Introduction: Despite adequate repair, coarctation of the aorta (CoA) with concomitant aortic arch hypoplasia (AAH) is associated with an increased risk of hypertension. However, the mechanism(s) involved in the development of hypertension are unknown
Hypothesis: We hypothesized that the aortic arch of infants with CoA + AAH would have differences in gene expression compared to infants with CoA alone, providing insight into the pathophysiologic mechanisms leading to later hypertension.
Methods: In 14 infants (7-CoA+AAH, 7-CoA alone) we analyzed mRNA after isolating the aortic tissue from the transverse aortic arch and the aorta distal to the CoA. An Affymetrix 1.0 genome array was performed to elucidate differences in gene expression between aortic arch and distal aorta from infants with CoA+AAH vs. CoA alone. RT-PCR validated genetic differences after quantifying the fold difference in expression by normalizing the aortic arch mRNA to the distal aorta.
Results: The micro-array data identified 807 genes that were significantly expressed from within the aortic arch of infants with CoA+AAH, predominantly in vascular smooth muscle cell regulation, cell division, and development. In contrast, infants with isolated CoA without AAH, demonstrated decreased number, and different focus, of gene expression (532 genes predominately involved in cell division). RT-PCR confirmed that aortic arch hepsin expression, a gene involved in cell growth, was significantly over-expressed in infants with CoA+AAH (26.1±18 vs. 0.5±10; p=0.05).
Conclusions: Infants with CoA+AAH have differences in aortic arch gene expression compared to infants with CoA alone. We postulate that these genetic variations contribute to the pathophysiologic changes that lead to later hypertension.
Article Information
vol. 130 no. Suppl 2 A18637
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Surgery, Univ of Rochester, Rochester, NY
- 2Pediatrics, Univ of Rochester, Rochester, NY
Abstract 17900: Sarcomere Gene Mutations in Left Ventricular Noncompaction
Asami Takasaki, Yukiko Hata, Keiichi Hirono, Nakaoka Hideyuki, Keijiro Ibuki, Sayaka Ozawa, Naoki Yoshimura, Naoki Nishida, Fukiko Ichida
Circulation. 2014;130:A17900
Abstract
Introduction: Left ventricular noncompaction (LVNC) is a primary cardiomyopathy with heterogeneous genetic origins, however, the genetic basis of disease in a large proportion of patients with LVNC is still unresolved. We evaluated the potential clinical impact of genetic analysis of sarcomere genes in patients with LVNC.
Methods: We investigated 93 Japanese LVNC patients, including 23 familial cases, for mutations of genes encoded sarcomeric proteins including myosin binding protein C (MYBPC3), β-myosin heavy chain (MYH7), α-tropomyosin (TPM1), cardiac troponin T (TNNT2), α-cardiac actin (ACTC) and cardiac troponin I (TNNI3). Of these 49 were infants and 44 were juvenile cases.
Results: We identified 28 sarcomeric gene mutations in 32 patients (34%) including 17 infants and 15 juvenile cases . These mutations were distributed among 5 genes, 12 in MYH7 and 9 in MYBPC3, 3 in TPM1, 2 in TNNT2, and 2 in ACTC1. Nineteen (68%) of the mutations were novel, affected conserved amino acid residues and were predicted to alter the structure of the proteins by in silico analysis. MYH7 and MYBPC3were the most prevalent disease genes and accounted for 81% of cases with mutation. Of note, 2 infants and 2 juvenile cases were compound or double heterozygous for 2 different mutations, and showed the most severe phenotype. Although most of the infants had clinical signs or symptoms of heart failure at initial presentation (88%), the majorities of juvenile cases were asymptomatic and identified only when screened for cardiac abnormalities, such as ECG screening (57%). Comparing sarcomere mutation-positive and mutation-negative LVNC probands showed no significant differences with respect to clinical characteristics at baseline and mortality during follow up both in infants and juvenile cases.
Conclusions: Mutations in sarcomere genes account for a significant proportion of patients with LVNC both in infants and juvenile cases. High incidence of novel mutations supports the concept that LVNC is part of a diverse spectrum of cardiac morphologies triggered by sarcomere protein gene defects.
Article Information
vol. 130 no. Suppl 2 A17900
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Asami Takasaki1;
- Yukiko Hata2;
- Keiichi Hirono1;
- Nakaoka Hideyuki1;
- Keijiro Ibuki1;
- Sayaka Ozawa1;
- Naoki Yoshimura3;
- Naoki Nishida2;
- Fukiko Ichida1
- 1Pediatrics, Univ of Toyama, Toyama, Japan
- 2Legal Medicine, Univ of Toyama, Toyama, Japan
- 3Ist department of Surgery, Univ of Toyama, Toyama, Japan
Abstract 16564: Analysis of the Mechanisms of Intravenous Immunoglobulin-Resistant Kawasaki Disease Using iPS Cell Technology
Kazuyuki Ikeda, Tomonaga Ameku, Yui Nomiya, Masahiro Nakamura, Satoshi Matsui, Tomoyo Yahata, Akiko Okamoto-Hamaoka, Chinatsu Suzuki, Yuki Kuchitsu, Akira Watanabe, Kenji Osafune, Kenji Hamaoka
Circulation. 2014;130:A16564
Abstract
Introduction: Kawasaki disease (KD) is a systemic vasculitis of unknown origin. Although the treatment of intravenous immunoglobulin (IVIG) significantly resolves inflammation, 10-20% of KD patients have persistent or recurrent fever after the administration of IVIG, and IVIG-resistant patients have a particularly high risk of developing coronary artery abnormalities.
Hypothesis: The mechanisms of IVIG-resistant KD have been analyzed using the patients’ leukocyte samples. However, vascular endothelial cells (ECs), closely related to the vasculitis of KD, have not been examined in the previous reports. We propose a hypothesis that ECs are mainly involved in the etiology of IVIG-resistance.
Methods: The purpose of this study is to establish new in vitro disease models of vasculitis using induced pluripotent stem cell (iPSC) technology, and clarify the mechanisms of IVIG-resistance in KD. Dermal fibroblasts or T cells from 2 IVIG-resistant and 2 IVIG-responsive KD patients were reprogrammed by episomal vectors encoding Oct3/4, Sox2, Klf4, L-Myc, LIN28, and p53 shRNA. The iPSC lines were then differentiated into ECs by using a previously-reported differentiation method, and the EC samples were subjected to the microarray analyses.
Results: The KD patient-derived iPSCs could be differentiated into ECs. The gene expression profiles were compared between iPS-derived ECs (iPS-ECs) generated from IVIG-resistant and IVIG-responsive KD patients. We found that 107 genes were at least two fold up-regulated and 101 genes were at least two fold down-regulated in iPS-ECs from IVIG-resistant KD patients compared with those from IVIG-responsive patients. The Principle Component Analysis (PCA) was performed, but the gene expression levels showed no significant differences between the groups. The Gene Set Enrichment Analysis (GSEA) revealed that the gene sets related to IL-6, NRAS (a member of the RAS oncogene family) and breast cancer were up-regulated in iPS-ECs from IVIG-resistant KD patients.
Conclusions: Taking into account that the concentration of IL-6 has been reported to be elevated in acute phase of IVIG-resistant KD, our results suggest that the up-regulation of IL-6 related genes in ECs might be involved in the pathogenesis of IVIG-resistant KD.
Article Information
vol. 130 no. Suppl 2 A16564
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Kazuyuki Ikeda1;
- Tomonaga Ameku2;
- Yui Nomiya2;
- Masahiro Nakamura2;
- Satoshi Matsui2;
- Tomoyo Yahata1;
- Akiko Okamoto-Hamaoka1;
- Chinatsu Suzuki1;
- Yuki Kuchitsu1;
- Akira Watanabe2;
- Kenji Osafune2;
- Kenji Hamaoka1
- 1Dept of Pediatric Cardiology and Nephrology, Kyoto Prefectural Univ of Medicine, Kyoto, Japan
- 2Cntr for iPS Cell Rsch and Application (CiRA), Kyoto Univ, Kyoto, Japan
Abstract 16646: Successful Expansion and Bioscaffolding of Clonogenic Cardiac Pericyte-Like Cells From Infants Affected by Congenital Heart Disease
Elisa Avolio, Iker Rodriguez-Arabaolaza, Somi Idowu, Jonathan Rowlinson, Helen Spencer, Federica Riu, Sadie Slater, Atsuhiko Oikawa, Mohamed Ghorbel, Massimo Caputo, Paolo Madeddu
Circulation. 2014;130:A16646
Abstract
Introduction: Current prostheses for correction of congenital heart disease (CHD) are unable to match the growth of an infant’s heart and deteriorate due to matrix degradation. Biological scaffolds integrated with progenitor cells able to grow and renew the prosthetic matrix may provide durable correction of CHD. We investigate the plasticity of cardiac pericyte-like cells obtained from CHD infants and their compatibility with a clinically certified prosthetic graft (CorMatrix).
Methods&Results: CD34+ CD31- Pericytes (CPs) were immuno-sorted from myocardial specimen leftovers (n=13) of neonates and infants undergoing repairs of CHD. CPs were expanded and characterized for surface antigens, plasticity toward cardiovascular lineages, clonogenicity and ability to colonize a CorMatrix patch. We successfully expanded CPs in culture for several passages to reach a high number of cells (>20 million at P5). Flow cytometry of expanded cells at P4 (n=7) indicates a mesenchymal phenotype (CD105, CD44, CD90) but very low expression of endothelial and hematopoietic markers. Fluorescent microscopy of CP lines (n=6) show abundant expression of pericyte (NG2, vimentin, PDGFR-β) and stemness markers (Oct-4, SOX-2, Nanog). By single-cell sorting, we demonstrated the clonogenic capacity of CPs. When cultured with differentiation media, CPs failed to acquire mature endothelial proteins (VEGFR2, vWF, VE-Cadherin), while they acquired markers of synthetic VSMCs (α-SMA, Calponin, non-muscle myosin B, RBP1). Moreover, when grown under cardiomyocyte inductive conditions, CPs acquired TBX5, CACNA1C and Connexin43 at the transcriptional level. Finally, we succeed in growing CPs on a CorMatrix membrane. Fluorescent microscopy showed vimentin-positive CPs not only at the surface, but also within the patch, indicating ability to colonize, grow and form a cellularized scaffold.
Conclusions: We show for the first time the possibility of expanding a clonogenic population of bona fide pericytes from hearts of CHD patients, differentiating them in cardiovascular cells, and growing them within extracellular matrices that are currently used in cardiac surgery. These data open new avenues for cellular functionalization of prosthetic material to correct CHD.
Article Information
vol. 130 no. Suppl 2 A16646
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Elisa Avolio;
- Iker Rodriguez-Arabaolaza;
- Somi Idowu;
- Jonathan Rowlinson;
- Helen Spencer;
- Federica Riu;
- Sadie Slater;
- Atsuhiko Oikawa;
- Mohamed Ghorbel;
- Massimo Caputo;
- Paolo Madeddu
- Sch of Clinical Sciences, Univ of Bristol, Bristol, United Kingdom
Abstract 16396: Phosphodiesterase-5 Expression and Activity is Increased in Children With Single Ventricle Heart Failure
Shelley D Miyamoto, Penny Nelson, Rebecca Sobus, Karin Nunley, Valencia Peterson, Brian L Stauffer, Carmen C Sucharov
Circulation. 2014;130:A16396
Abstract
Introduction: Single ventricle congenital heart disease (SV) is the leading cause of cardiovascular death and indication for heart transplantation in infancy. There are no proven therapies for SV heart failure (HF). Human and animal models of HF demonstrate that myocardial phosphodiesterase-5 (PDE5) is increased with cardiac stress and treatment with a PDE5 inhibitor (PDE5i) results in enhanced cardiac function and prevents remodeling. Sildenafil, a PDE5i, is increasingly utilized for the treatment of patients suffering from failing SV. The objective of this study was to determine myocardial PDE5 expression and activity in children transplanted for failing SV.
Methods: At the time of cardiac transplantation, explanted pediatric hearts were immediately cooled in ice cold oxygenated Tyrodes in the operating room. The tissue is rapidly dissected, flash frozen and stored at -800C until further use. RNA, protein and cytosolic fractions were isolated from explanted right ventricle (RV) tissue from SV and non-failing (NF) donors. RTqPCR for PDE5, Western blot (normalized to calnexin loading control) for PDE5 and PDE5 activity assays were performed. For PDE 5 activity, cGMP hydrolysis was measured using [3H]cGMP as the substrate. Sildenafil was added to measure PDE5 specific activity, and activity was calculated using nonlinear regression.
Results: All patients used in the SV analysis had a failing morphologic RV and were selected from a cohort of 17 SV (median age 0.5, range 0.05-10 yrs) and 8 NF controls (median age 7, range 1.3-13 yrs). PDE5 gene expression was higher in SV myocardium compared to NF (1.9±0.7, n=17 SV vs 1.1±0.5 ct fold change, n=8 NF, p=0.02). There was a trend towards higher PDE5 protein expression in SV myocardium compared to NF (1.5±0.7, n=4 SV vs 1.0±0.4 protein expression normalized to NF, n=3 NF; p=ns). There was increased PDE5 activity in SV compared to NF (22.3±1.2, n=3 SV vs 11.9±4.2 pmol/mg/min, n=2 NF; p=0.02).
Conclusions: There is increasing evidence that PDE5i has beneficial direct myocardial effects. There is increased PDE5 gene expression and activity in failing SV myocardium compared to NF control suggesting that PDE5 may represent a promising therapeutic target in this challenging population.
Article Information
vol. 130 no. Suppl 2 A16396
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Shelley D Miyamoto1;
- Penny Nelson2;
- Rebecca Sobus2;
- Karin Nunley1;
- Valencia Peterson2;
- Brian L Stauffer2;
- Carmen C Sucharov2
- 1Pediatric Cardiology, Univ of Colorado Denver, Children’s Hosp Colorado, Aurora, CO
- 2Cardiology, Univ of Colorado Denver, Aurora, CO
Abstract 11840: Disease-Specific Induced Pluripotent Stem Cells Identify the Transcriptional Repression and Epigenetic Modification of NKX2-5, HAND1, and NOTCH1 During Cardiac Development of Hypoplastic Left Heart Syndrome
Junko Kobayashi, Masashi Yoshida, Suguru Tarui, Shuta Ishigami, Michihiro Okuyama, Yusuke Nagai, Shingo Kasahara, Keiji Naruse, Hiroshi Ito, Shunji Sano, Hidemasa Oh
Circulation. 2014;130:A11840
Abstract
Backgrounds: Although heterozygous mutations have been reported in patients with hypoplastic left heart syndrome (HLHS), no genes have been found to specifically cause this syndrome. The aim of this study is to investigate the complex network of transcriptional regulation and epigenetic modification during the cardiac development of HLHS by using patient-specific induced pluripotent stem (iPS) cells.
Methods: Cardiac progenitor cells (CPCs) were isolated and four-independent iPS cell lines were generated from HLHS- and bi-ventricular (BV) heart-derived CPCs. Real-Time RT-PCR, exome sequencing, ChIP assay, and luciferase reporter assay were performed and compared with human embryonic stem and 201B7 iPS cells as controls.
Results: HLHS-derived iPS cells could give rise to cardiomyocytes with significantly lower expression of cardiac troponin-T (TNNT2), NKX2-5, HAND1/2, NOTCH1-HEY1/2 and TBX2 when compared with those from 201B7 and BV-derived iPS cells. To determine the target transcripts responsible for cardiac development of HLHS, luciferase reporter analyses of serum response element (SRE), TNNT2, and natriuretic peptide A (NPPA) were examined. We found that transcriptional activation of SRE, TNNT2, and NPPA was significantly suppressed in HLHS-derived CPCs and iPS cells compared with those from BV heart. No mutations in NKX2-5, HAND1, and NOTCH1 were identified in patients analyzed; however, repressed promoter activities of SRE and TNNT2 in HLHS-derived CPCs and iPS cells could be fully restored by transient transfection of these three transcripts by synergetic fashion. Notably, the transcriptional activation of NPPA was controlled by NKX2-5-dependent mechanism and loss-of-function studies by shRNA confirmed these observations. In addition, ChIP assay showed a marked decrease in histone enhancer marks such as H3K4me2 and acH3 but gain of repressive mark H3K27me3 on NKX2-5 promoter in differentiated HLHS-derived iPS cells.
Conclusions: These findings suggest that patient-specific iPS cells may provide molecular insights into complex transcriptional and epigenetic mechanisms, at least in part, through combinatorial expression of NKX2-5, HAND1, and NOTCH1 that coordinately contribute to cardiac malformations in HLHS.
Article Information
vol. 130 no. Suppl 2 A11840
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Junko Kobayashi1;
- Masashi Yoshida2;
- Suguru Tarui1;
- Shuta Ishigami1;
- Michihiro Okuyama1;
- Yusuke Nagai3;
- Shingo Kasahara1;
- Keiji Naruse3;
- Hiroshi Ito2;
- Shunji Sano1;
- Hidemasa Oh4
- 1Cardiovascular Surgery, Okayama Univ, Okayama, Japan
- 2Cardiovascular Medicine, Okayama Univ, Okayama, Japan
- 3Cardiovascular Physiology, Okayama Univ, Okayama, Japan
- 4Cntr for Innovative Clinical Medicine, Okayama Univ Hosp, Okayama, Japan
Abstract 17861: Identification of Cardiovascular Lineage Descendants at Single Cell Resolution
Guang Li, Karolina Plonowska, Rajarajan Kuppusamy, Sean M. Wu
Circulation. 2014;130:A17861
Abstract
BACKGROUND: The transcriptional profile of cardiac cells derived from murine embryos and from mouse embryonic stem cells (mESCs) has primarily been studied as a cell population. However, the characterization of gene expression in these cells at a single cell level may demonstrate unique variations not appreciated as a pool.
METHODS AND RESULTS: To establish a single cell quantitative PCR platform and perform side-by-side comparison between cardiac progenitors cells (CPCs) and cardiomyocytes (CMs) derived from our previously described Nkx2.5 enhancer-eGFP mESC and mouse embryos, we generated a reference map for single cardiovascular cells through quantifying lineage-defining genes for CPCs, CMs, smooth muscle cells (SMCs), endothelial cells (EDCs), fibroblasts, and mESCs using the Fluidigm microfluidic-enabled multiplex PCR assay. This panel was then applied against day 10.5 embryonic heart single cells to demonstrate its ability to identify chamber-specific CMs, endocardial cells, and fibroblasts. In addition, we compared the gene expression profiles of Nkx2.5 enhancer-eGFP embryo- and mESC-derived CPCs and CMs at different developmental stages and showed that single mESC-derived CM is transcriptionally similar to embryo-derived CM up to the neonatal stage. Furthermore, we show that time-lapse microscopy coupled with single cell expression assay can resolve the identity and the lineage relationship of progenies of single cultured CPC. With this approach, we found differential propensity for mESC-derived CPCs to become SMCs and CMs, whereas embryo-derived CPC to become CMs or EDCs.
CONCLUSIONS: Our single cell expression profiling assays demonstrate the transcriptional similarity between mESC and embryo-derived CPC and CM up to the neonatal stage of development as well as differences in the propensity of CPCs to differentiate into cardiovascular cells. Single cell expression analysis appears to be a powerful tool to address the unique behavior of individual embryo- or mESC-derived cardiovascular cells.
Article Information
vol. 130 no. Suppl 2 A17861
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Guang Li;
- Karolina Plonowska;
- Rajarajan Kuppusamy;
- Sean M. Wu
- Stanford, Cardiovascular Institute, Stanford, CA
Abstract 16771: Inferring Dynamic Gene-microRNA Regulatory Networks in Cardiac Differentiation by Integrating Multi-Dimensional Data
Wuming Gong, Naoko Koyano-Nakagawa, Daniel J Garry
Circulation. 2014;130:A16771
Abstract
Background: Decoding the temporal control of gene and microRNA expression patterns are key to understanding the complex mechanism of developmental decisions during heart development. High-throughput methods have been employed to systematically study the dynamic and coordinated nature of cardiac differentiation at the global level with multiple dimensions. There is pressing need to develop systems way to integrate these data from individual studies and infer the dynamic regulatory networks in an unbiased fashion.
Methods: We developed a two-step strategy to integrate data from (1) temporal RNA-seq, (2) temporal histone modifications ChIP-seq, (3) temporal microRNA microarray, (4) microRNA target sites, (5) transcription factor (TF) ChIP-seq and (6) gene perturbation, to reconstruct the dynamic network. First, we trained a logistic regression model to predict the probability (LR score) of any base being bound by 543 TFs with known positional weight matrices. Second, six dimensions of data were combined by time-varying dynamic Bayesian network model to infer the dynamic networks. Our method not only infers the time-varying networks between different stages of heart development, but also identifies the TF binding sites.
Results: The LR scores of known ESC and heart enhancers were significantly higher than random regions (p <10-100), suggesting that a high LR score is a reliable indicator for functional TF binding sites. Our network inference model identified a region with elevated LR score approximately -9400 bp upstream of the transcriptional start site of Nkx2-5, which overlapped with a previously reported enhancer region (-9435 to -8922 bp). TFs such as Tead1, Gata4, Msx2, and Tgif1 were predicted to bind to this region and participate in the regulation of Nkx2-5. Our model also predicted 435 significant TF-microRNA-gene network motifs that may be important for the differentiation from cardiac progenitors to cardiomyocytes.
Conclusions: We report a novel method to systematically integrate multi-dimensional omics data and reconstruct the gene-microRNA regulatory networks. This method will allow one to rapidly determine the cis-modules and TF-microRNA-gene network motifs that regulate key genes during cardiac differentiation.
Article Information
vol. 130 no. Suppl 2 A16771
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Wuming Gong;
- Naoko Koyano-Nakagawa;
- Daniel J Garry
- Dept of Medicine, Univ of Minnesota, Minneapolis, MN
Abstract 15973: miRNA-Hippo Pathway Plays a Critical Role in the Cardiac Conduction System
Jun Wang, Sylvia Evans, James Martin
Circulation. 2014;130:A15973
Abstract
The cardiac conduction system (CCS) is required for initiating and maintaining regular rhythmic heartbeats and the CCS defects can give rise to cardiac arrhythmia, a leading cause for morbidity worldwide. Given the poor self-repair potential in the adult human CCS, it is critical to elucidate the molecular mechanisms limiting the CCS regeneration to facilitate developing efficient cardiovascular therapies. MicroRNAs (miRs) are small non-coding RNAs that repress gene expression post-transcriptionally. The miR-17-92 cluster can induce cardiomyocyte proliferation and regeneration. Hippo signaling, an ancient organ size control pathway, represses cardiomyocyte proliferation and regeneration. Here we found that both miR-17-92 and Hippo signaling were active in the CCS. Specific disruption of either miR-17-92 or Hippo signaling in the CCS gave rise to cardiac arrhythmias in mice. Notably, miR-17-92 regulates Hippo signaling through directly repressing Lats2, a core Hippo pathway component. In miR-17-92 null mutant hearts, up-regulated Lats2 led to increased Hippo pathway activity. Moreover, we performed chromatin immunoprecipitation deep sequencing (ChIP-Seq) using Yap antibody, the Hippo signaling effector, which data suggested that Hippo signaling regulates genes involved in the CCS homeostasis. Together, our data indicate a novel miR-Hippo genetic pathway plays critical function in the CCS.
Article Information
vol. 130 no. Suppl 2 A15973
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Molecular Physiology and Biophysics, Baylor College of Medicine, Houston, TX
- 2Dept of Medicine, Univ of California San Diego, La Jolla, CA
Abstract 15657: Regulation of Cardiac Regeneration by Hippo Pathway and Dystrophin Glycoprotein Complex
Yuka Morikawa, James F Martin
Circulation. 2014;130:A15657
Abstract
Regeneration of the mammalian heart is limited in adults. In rodents, endogenous regenerative capacity exists during development and in neonate but is rapidly repressed after birth. We are elucidating the mechanisms responsible for regenerative repression and applying this knowledge to reactivate cardiac regeneration in adult hearts. We have previously shown that the Hippo pathway is responsive for regenerative repression, however, the molecular and cellular mechanism responsible remain unclear.
The Hippo pathway controls heart size by repressing myocardial cell proliferation during development. By deleting Salv, a modulator of Hippo pathway, we found myocardial damage in the postnatal and adult heart was repaired anatomically and functionally. This heart repair occurred primarily through proliferation of preexisting cardiomyocyte. We observed that cardiomyocytes in border the zone protrude and fill the damage area during Hippo-mediated cardiac regeneration and thus preventing formation of fibrotic scars. The molecular analysis identified components of dystrophin glycoprotein complex (DGC) as downstream targets of Hippo pathway. The DGC anchors the cytoskeleton and extracellular matrix and is involved in cell migration. The studies using the muscular dystrophy mouse model, mdx, reveals that DGC is required for endogenous cardiac regeneration and cardiomyocyte protrusion. Taken together, we show that cardiomyocyte protrusion is an essential process for cardiac regeneration and the Hippo pathway regulates it through regulating DGC. Our studies provide insights into the mechanisms leading to repair of damaged hearts from endogenous cardiomyocytes and novel information into DGC function.
Article Information
vol. 130 no. Suppl 2 A15657
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Cardiomyocyte Renewal, Texas Heart Institute, Houston, TX
- 2Molecular Physiology, Baylor College of Medicine, Houston, TX
Abstract 14000: miR-322/503 Cluster Drives Cardiac Differentiation by Inhibiting CUG-binding Protein 1
Xiaopeng Shen, Ashley Benham, Benjamin Soibam, Wei Yu, Robert J Schwartz, Yu Liu
Circulation. 2014;130:A14000
Abstract
Understanding the mechanisms of early cardiac fate determination may lead to better approaches in promoting heart regeneration after injury. MicroRNAs (miRNAs) involved in the process are particularly interesting due to their small profile and relatively shorter path to clinic. With Mesp1 as the marker, we used a Mesp1-Cre/Rosa-EYFP reporter system to track the earliest cardiac progenitors, and identified the miRNAs enriched in these cells. Among them, the miR-322/503 cluster is found to be a powerful regulator of the cardiac program: (1) in a screening of more than 20 CPC-enriched miRNAs, miR-322/503 was the most powerful in driving calcium flux activity in mouse embryonic stem cells (mESCs) differentiation; (2) induced ectopic expression of miR-322/503 to mimic the natural course in mESCs led to α-actinin expression and significant increases of cardiac transcription factors (Tbx5, Mef2C, Nkx2-5 and α-MHC); (3) inhibitors of miR-322 and miR-503 significantly reduced expression of α-actinin and the above cardiac TFs. Remarkably, miR-322/503 regulates the cardiac program by inhibiting an RNA-alternative splicing/decay factor, CUG-binding protein 1 (Celf1), which is also known for a role in myotonic dystrophy pathogenesis. The evidences include (i) miR-322 and miR-503 had a shared target site at 3’UTR of Celf1; (ii) expression patterns of miR-322/503 and Celf1 were mutually exclusive, with the highest Celf1 expression in the brain; (iii) miR-322/503 repressed Celf1 protein expression in a dose-dependent manner; (iv) Celf1-shRNA induced up-regulation of cardiac transcription factors and α-actinin, mimicking the function of miR-322/503; (v) ectopic Celf1 expression repressed expression of cardiac transcription factors, while promoted expressions of early neural markers, including Sox1, Zic1, Nestin and Pax6. In summary, we have identified a miR-322/503-Celf1 pathway that promotes cardiac differentiation by preventing activation of other lineages. This new regulatory mechanism may be used to direct cardiac regeneration after heart injury, and treat myotonic dystrophy where Celf1 up-regulation is responsible for skeletal muscle wasting and other symptoms.
Article Information
vol. 130 no. Suppl 2 A14000
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Xiaopeng Shen;
- Ashley Benham;
- Benjamin Soibam;
- Wei Yu;
- Robert J Schwartz;
- Yu Liu
- Biology&Biochemistry, Univ of Houston, Houston, TX
Abstract 18522: Exome Chip Analysis From the CASPER Registry Highlights Potential Genetic Variant Associated With Unexplained Cardiac Arrest
Rafik Tadros, Andrew D Krahn, Nathalie Chami, Jeffrey S Healey, Vijay S Chauhan, David H Birnie, Jean Champagne, Shubhayan Sanatani, Paul Angaran, Robert M Gow, Jean-Claude Tardif, John D Rioux, Marie-Pierre Dube, Simon de Denus, Santabhanu Chakrabarti, Brenda Gerull, Laurence Sterns, Raymond Yee, Lorne J Gula, George J Klein, Michael H Gollob, Martin Gardner, Christopher S Simpson, Mario Talajic, Guillaume Lettre
Circulation. 2014;130:A18522
Abstract
Introduction: The genetics of unexplained cardiac arrest (UCA) remains largely elusive. We sought to identify genetic variants linked to UCA in a case-control genetic association study.
Methods: Cases are UCA probands from the Cardiac Arrest Survivors With Preserved Ejection Fraction Registry (CASPER), which included individuals with a history of UCA or sudden cardiac death (SCD) with normal autopsy. Exclusion criteria were coronary/structural heart diseases, or a baseline ECG with type I Brugada or prolonged QTc (males>460ms, females>480ms). Controls were individuals without coronary/structural heart diseases, ventricular arrhythmia or family history of SCD, from the Montreal Heart Institute Biobank. We performed exome-wide genotyping using the Illumina HumanExome BeadChip (>250,000 mostly coding markers). Each case was matched with 3 controls from a 3139 individuals dataset using identity-by-descent. Association analysis was performed using the Cochran-Mantel-Haenszel test, controlling for sex. We validated genotyping of most significant variants using Sequenom technology.
Results: After quality checks, we compared allele frequencies at 76157 polymorphic markers in 79 cases and 237 matched controls. Genotyping success rate was 99% in both groups. We identified 17 variants with P-values <1×10-4, whereas we would have expected 7 under the null hypothesis of no association. This reflects an enrichment of variants associated with UCA. The strongest association with UCA risk was observed at a single nucleotide polymorphism (SNP) in POLRMT. Minor allele frequency (MAF) in cases and controls was 0.048 and 0.011, respectively (p = 9.7×10-7, odds ratio = 5.0). MAF in controls was similar to those reported in Exome Sequence Project (0.014) and 1000 Genomes (0.007). POLRMT encodes a mitochondrial DNA-directed RNA polymerase. Interestingly, POLRMT is located next to HCN2, which encodes an ion channel previously implicated in ventricular arrhythmia.
Conclusion: In this case-control study from the CASPER registry, a SNP in POLMRT is strongly associated with UCA. Replication studies are ongoing. If the association is confirmed, the mechanism of arrhythmogenesis might involve deregulation of gene expression of a nearby ion channel gene.
Article Information
vol. 130 no. Suppl 2 A18522
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Rafik Tadros1;
- Andrew D Krahn2;
- Nathalie Chami3;
- Jeffrey S Healey4;
- Vijay S Chauhan5;
- David H Birnie6;
- Jean Champagne7;
- Shubhayan Sanatani8;
- Paul Angaran9;
- Robert M Gow10;
- Jean-Claude Tardif3;
- John D Rioux3;
- Marie-Pierre Dube3;
- Simon de Denus3;
- Santabhanu Chakrabarti2;
- Brenda Gerull11;
- Laurence Sterns12;
- Raymond Yee13;
- Lorne J Gula13;
- George J Klein13;
- Michael H Gollob14;
- Martin Gardner15;
- Christopher S Simpson16;
- Mario Talajic1;
- Guillaume Lettre3
- 1Dept of Medicine, Montreal Heart Institute, Montreal, Canada
- 2Div of Cardiology, Univ of British Columbia, Vancouver, Canada
- 3Rsch Cntr, Montreal Heart Institute, Montreal, Canada
- 4Population Health Rsch Institute, McMaster Univ, Hamilton, Canada
- 5Peter Munk Cardiac Cntr, Univ Health Network, Toronto, Canada
- 6Arrhythmia Service, Univ of Ottawa Heart Institute, Ottawa, Canada
- 7Cardiology, Quebec Heart and Lung Institute, Quebec, Canada
- 8Cardiac Sciences Program, BC Children’s Hosp, Vancouver, Canada
- 9Cardiac Arrhythmia Service, St Michael’s Hosp, Toronto, Canada
- 10Div of Cardiology, Children’s Hosp of Eastern Ontario, Ottawa, Canada
- 11Dept of Cardiac Sciences, Univ of Calgary, Calgary, Canada
- 12Cardiology, Royal Jubilee Hosp, Victoria, Canada
- 13Div of Cardiology, Western Univ, London, Canada
- 14Div of Cardiology, Univ of Ottawa Heart Institute, Ottawa, Canada
- 15Div of Cardiology, QEII Health Science Cntr, Halifax, Canada
- 16Div of Cardiology, Queen’s Univ, Kingston, Canada
Abstract 18543: Whole Exome Sequencing in Sudden Infant Death Syndrome Identifies a High Proportion of Putative Pathogenic and Functionally Significant Rare Variants Related to Inherited Cardiac Conditions
Leonie C Wong, Michael A Simpson, David J Tester, David R FitzPatrick, Jacob Tfelt-Hansen, Steve Bevan, James S Ware, Margaret J Evans, Peter J Fleming, Craig C Platt, Iona J Jeffrey, Marta C Cohen, Michael J Ackerman, Elijah R Behr
Circulation. 2014;130:A18543
Abstract
Introduction: Sudden Infant Death Syndrome (SIDS) is defined as the sudden death of an infant which remains unexplained after comprehensive clinical and pathological assessment. Prior studies have implicated inherited cardiac conditions (ICC).
Hypothesis: Whole exome sequencing will identify a significant burden of putative pathogenic (PP) and rare functionally significant (RFS) cardiac genetic variants in SIDS.
Methods: 96 SIDS cases with complete phenotypic data were selected from 434 cases. Prone sleeping position was excluded to enrich for genetic risk. Genomic DNA underwent target enrichment using Agilent SureSelect Human All Exon v5 kit and sequencing on Illumina HiSeq. Reads were aligned to GRCh37 reference genome with Novoalign and variants called with SAMtools. Principal component analysis identified 82 cases of North European (NE) ancestry. Variants were annotated with Annovar and compared to control data from a local exome database, 1000 Genomes and ESP. Analysis focused on 83 known ICC susceptibility genes (27 channelopathy-associated [CH]; 56 cardiomyopathy-associated [CM]). PP was defined as ultra rare nonsense or frameshift mutations or other variants previously reported as disease-associated and absent in controls. RFS was defined as variants with abnormal functional characterization and <0.5% prevalence in controls.
Results: 20/82 cases (24%) carried at least 1 PP/RFS variant: 12 CH variants in 9 genes (AKAP9, ANK2, CACNB2, CAV3, KCNE2, RYR2, SCN5A, SNTA1, TRDN) in 13 cases; 7 CM variants in 5 genes (ABCC9, ACTN2, CSRP3, TCAP, TTN) in 8 cases. Variants were most commonly identified in the 3-6 months age group (p=0.009). There were no other significant phenotypic differences between variant-positive and negative cases (Table 1).
Conclusions: There was an 11% yield of PP variants in non-prone sleeping SIDS cases of NE ancestry, with an additional 13% hosting RFS variants. These may provide a potential substrate in the pathophysiology of SIDS.
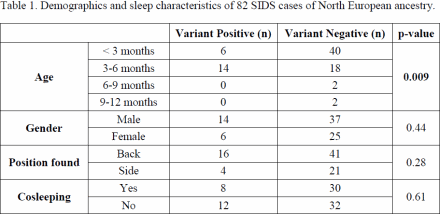
Article Information
vol. 130 no. Suppl 2 A18543
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Leonie C Wong1;
- Michael A Simpson2;
- David J Tester3;
- David R FitzPatrick4;
- Jacob Tfelt-Hansen5;
- Steve Bevan6;
- James S Ware7;
- Margaret J Evans8;
- Peter J Fleming9;
- Craig C Platt10;
- Iona J Jeffrey11;
- Marta C Cohen12;
- Michael J Ackerman3;
- Elijah R Behr1
- 1Cardiac and Cell Sciences Institute, St George’s Univ of London, London, United Kingdom
- 2Div of Genetics and Molecular Medicine, King’s College London, London, United Kingdom
- 3Dept of Internal Medicine, Div of Cardiovascular Diseases, Mayo Clinic, Rochester, MN
- 4MRC Human Genetics Unit, Univ of Edinburgh, Edinburgh, United Kingdom
- 5Dept of Cardiology, Copenhagen Univ Hosp Rigshospitalet, Copenhagen, Denmark
- 6Neurology Unit, Univ of Cambridge, Cambridge, United Kingdom
- 7NIHR Cardiovascular Biomedical Rsch Unit, Imperial College London, London, United Kingdom
- 8Dept of Pathology, Royal Infirmary of Edinburgh, Edinburgh, United Kingdom
- 9Cntr for Child and Adolescent Health, Univ of Bristol, Bristol, United Kingdom
- 10Dept of Cellular Pathology, Bristol Royal Infirmary, Bristol, United Kingdom
- 11Dept of Cellular Pathology, St George’s Hosp, London, United Kingdom
- 12Histopathology Dept, Sheffield Children’s NHS Foundation Trust, Sheffield, United Kingdom
Abstract 17982: A lamin A/C Synonymous Mutation Creates a Novel Splice Site and Causes Progressive Atrioventricular Conduction Defect
Kaoru Ito, Barbara McDonough, Joshua M Gorham, Steven R DeParma, Emily E Adler, Syed M Mohiuddin, Diane Fatkin, J G Seidman, Christine E Seidman
Circulation. 2014;130:A17982
Abstract
Background: Mutations in the lamin A/C, encoded by LMNA, produce diverse genetic disorders, collectively termed laminopathies including disease of cardiac and skeletal muscles, peripheral neuropathy, lipodystrophy and premature aging syndromes. Cardiac phenotypes associated with LMNA mutations include progressive cardiac conduction defects and dilated cardiomyopathy (DCM), and this has been attributed predominantly to missense variants and less commonly to truncating variants. Synonymous sequence variants are usually considered benign and have not been investigated in cardiac laminopathies. Here we report the identification of A LMNA synonymous mutation that mimics a splice donor consensus sequence and causes a 15 amino acid deletion in the exon 4.
Methods and Results: We conducted a linkage analysis by genome-wide SNP genotyping method and found one region with LOD score > 4 on chr1q21.3-q23.3, which includes LMNA. Exome sequencing was performed in 4 affected subjects. 3 unique variants that were shared by the affected Subjects were identified in the linkage region. One variant, a synonymous change in the LMNA gene, was selected for further analysis, since LMNA is a strong candidate gene for the family’s phenotype and the variant was predicted to introduce a new potential splice donor site. Sanger sequencing samples from the extended pedigree confirmed that the synonymous LMNA variant was present in all affected subjects and absent from unaffected subjects. Evaluation of myocardial tissue from one affected family member using RT-PCR showed the expected band of 240 bp size and an additional smaller band of 195 bp size, suggestive a potential effect of the variant on RNA splicing. Sanger sequencing of the smaller band revealed a 45-bp in-frame deletion in the tail of the exon4, indicating that this variant activates a cryptic splice donor site.
Conclusion: Here we report a novel synonymous LMNA variant, c.768G>A, in a family with DCM and conduction-system disease, this variant appears to alter RNA splicing by activating a cryptic splice donor site. These findings expand the spectrum of LMNA mutations associated with cardiac laminopathies and highlight the potential pathogenicity of synonymous sequence variants.
Article Information
vol. 130 no. Suppl 2 A17982
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Kaoru Ito1;
- Barbara McDonough1;
- Joshua M Gorham1;
- Steven R DeParma1;
- Emily E Adler1;
- Syed M Mohiuddin2;
- Diane Fatkin3;
- J G Seidman1;
- Christine E Seidman1
- 1Genetics, Harvard Med Sch, Boston, MA
- 2Div of Cardiology, Creighton Univ Sch of Medicine, Omaha, NE
- 3Cardiology, Victor Chang Cardiac Rsch Institute, Darlinghurst, Australia
Abstract 17517: Genetics of Left Ventricular Noncompaction Cardiomyopathy
Jan Haas, Zhu Feng, Christian Geier, Regina Pribe-Wolferts, Karen S Frese, Elham Kayvanpour, Farbod Sedaghat-Hamedani, Barbara Peil, Justo Lorenzo Bermejo, Jennifer Franke, Andreas Keller, Yuhua Liao, Hugo A Katus, Benjamin Meder
Circulation. 2014;130:A17517
Abstract
Left ventricular noncompaction cardiomyopathy (LVNC) is an increasingly recognized cause of heart failure, arrhythmias, thrombembolic events, and sudden cardiac death. To better understand the contribution of genetic factors to this disease, allowing a better diagnosis, counseling of affected families and estimation of prognosis, we have performed whole-exome deep-sequencing on a cohort of in 104 subjects (67 unrelated LVNC probands, 26 affected relatives form 15 families and 11 additional healthy relatives). By annotating the detected variants with mutation databases (HGMD), we were able to for the first time show an overlap between the distinct causes of LVNC and other genetic cardiomyopathies, as 8 mutations were previously reported to cause hypertrophic cardiomyopathy (HCM), 9 dilated cardiomyopathy (DCM) and 6 arrhythmogenic right ventricular cardiomyopathy (ARVC). Besides already known genetic causes, we identified a number of genes contributing to different pathways, so far not discussed to be of relevance for LVNC. From those, TTN truncating mutations were very frequent, affecting 17.9% of all index patients and one fifth of the familial cases. For two genes, which have already been linked to cardiac noncompaction in animal models, we are first to describe a relevant number of patients carrying mutations in these genes NCOR2 (frequency = 7.5%) and XIRP2 (4.5%). By performing segregation and linkage analyses, as well as in vivo studies, we provide for the first time evidence for a gene that was previously not known to cause a human disease, Compactin1. In conclusion, we show that LVNC has a substantial genetic component and suggest new candidate genes for a functional dissection of their contribution to cardiac development and as potential therapeutic target.
Article Information
vol. 130 no. Suppl 2 A17517
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Jan Haas1;
- Zhu Feng2;
- Christian Geier3;
- Regina Pribe-Wolferts1;
- Karen S Frese1;
- Elham Kayvanpour1;
- Farbod Sedaghat-Hamedani1;
- Barbara Peil4;
- Justo Lorenzo Bermejo4;
- Jennifer Franke1;
- Andreas Keller5;
- Yuhua Liao6;
- Hugo A Katus1;
- Benjamin Meder1
- 1Internal Medicine III, Univ Hosp Heidelberg, Heidelberg, Germany
- 2Internal Medicine IIIInstitute of Cardiology, Union Hosp, Tongji Med College, Huazhong Univ of Science and Technology, Wuhan, China
- 3Experimental and Clinical Rsch Cntr (ECRC), a joint cooperation of Charité Med Faculty and, Experimental and Clinical Rsch Cntr (ECRC), a joint cooperation of Charité Med Faculty and Max Delbrück Cntr for Molecular Medicine, Berlin, Germany
- 4Institute of Med Biometry and Informatics (IMBI), Univ Hosp Heidelberg, Heidelberg, Germany
- 5Dept of Bioinformatics, Univ of Saarland, Saarbrücken, Heidelberg, Germany
- 6Departement of cadiology, Institute of Cardiology, Union Hosp, Tongji Med College, Huazhong Univ of Science and Technology, Heidelberg, China
Abstract 15841: Whole Exome Sequencing and Analysis for Sudden Unexplained Death in the Young: A Case Series
Nupoor Narula, David J Tester, Anna Paulmichl, Joseph J Maleszewski, Michael J Ackerman
Circulation. 2014;130:A15841
Abstract
Introduction: Annually, thousands of sudden deaths in individuals under the age of 35 years remain unexplained following a medico-legal autopsy and are termed autopsy negative sudden unexplained death in the young (SUDY). Cardiomyopathies, channelopathies, and metabolic disorders may underlie a significant number of SUDY cases. Previously, we demonstrated that 25% of autopsy-negative SUDY cases had mutations in the 4 major cardiac ion channel genes (KCNQ1, KCNH2, SCN5A, and RYR2). However, over 100 sudden death-susceptibility genes have been discovered and may be implicated in SUDY.
Objective: We explored the utility of whole exome sequencing (WES) followed by gene-specific surveillance as an efficient and effective means of performing post-mortem genetic testing in SUDY.
Methods: Postmortem WES was performed on 14 consecutively-referred white SUDY victims (57% men; average age at death 17.4 ± 8.6 years) using the Agilent SureSelect Human All Exon V4+UTR capture kit and an Illumina HiSeq 2000 sequencer. Following variant alignment (hg19) and annotation, 117 cardiac channelopathy-, cardiomyopathy-, and metabolic disorder-susceptibility genes were surveyed to identify putative SUDY-associated mutations. Potentially pathogenic variants had to be non-synonymous and ultra-rare [i.e. absent in all 3 evaluated exome databases (1,000 Genome Project, the NHLBI GO Exome Sequencing Project, and Exome Chip Design)].
Results: On average, each SUDY case had 12,758 ± 2016 non-synonymous variants, of which 79 ± 15 localized to the 117 evaluated genes. Overall, 8 unique, ultra-rare variants (7 missense, 1 in-frame insertion) identified in 6 genes (3 in TTN; 1 each in CACNA1C, JPH2, MYH7, VCL, RYR2) were detected in 7 of 14 cases (50%). Of the 7 missense alterations, 2 (T171M-CACNA1C, I22160T-TTN) were predicted damaging by 3 in-silico tools.
Conclusions: Although WES and gene-specific surveillance is an efficient and effective strategy to detect rare, potentially lethal, genetic variants, the accurate interpretation of each variant is daunting. Importantly, rarity, even ultra-rarity, does not equal pathogenicity even when the ultra-rare variant resides within a so-called sudden death-susceptibility gene.
Article Information
vol. 130 no. Suppl 2 A15841
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Internal Medicine, Mayo Clinic, Rochester, MN
- 2Internal Medicine/Cardiovascular Diseases, Mayo Clinic, Rochester, MN
- 3Laboratory Medicine and Pathology, Mayo Clinic, Rochester, MN
Abstract 15882: Gene-Panel Based Next Generation Sequencing (NGS) Greatly Improves Clinical Genetic Diagnostics in Inherited Cardiomyopathies
Jan D Jongbloed, Anna Pósafalvi, Renée C Niessen, Yvonne M Hoedemaekers, Paul A van der Zwaag, Daniela Q Barge-Schaapveld, Sebastiaan R Piers, Jasper J van der Smagt, Folkert W Asselbergs, Rudolf A de Boer, Maarten P van den Berg, Rowida Almomani, Richard J Sinke, J. P van Tintelen
Circulation. 2014;130:A15882
Abstract
INTRODUCTION: NGS techniques can be successfully applied to find mutations underlying genetic cardiomyopathies. However, Exome Sequencing (ES) shows incomplete representation and coverage of exons, leading to clinically relevant mutations being missed. Thus, ES will, at least for now, coexist in clinical genetic diagnostics with other NGS-based strategies, such as gene-panel based resequencing. Therefore, we aimed at evaluating the yield of gene-panel based resequencing of 55 genes in cardiomyopathy patients referred to our department.
METHODS: We constructed an enrichment kit targeting 55 cardiomyopathy genes and implemented this into routine diagnostics. We evaluated our first 162 patients: 47 fulfilled generally accepted clinical criteria for hypertrophic cardiomyopathy (HCM), 72 fulfilled the Mestroni criteria for dilated cardiomyopathy (DCM) and 3 were diagnosed with arrhythmogenic cardiomyopathy (ACM). In addition, 23, 11 and 6 cases showed signs of DCM, HCM and ACM, yet did not fulfill the formal criteria. Additional cosegregation analyses were performed to further support pathogenicity of potentially causal mutations.
RESULTS: In the DCM cohort 40 pathogenic or likely pathogenic mutations were identified (55%; 40/72). Mutations in TTN were found in 14% of DCM patients (10/72). The yield in criteria positive HCM and ACM patients was 40% (17/43) and 33% (1/3). In patients not fully fulfilling criteria for DCM, HCM and ACM the yield was 52% (12/23), 36% (4/11) and 83% (5/6). In 13 % (21/162) of cases two or more (potentially) pathogenic mutations were identified. Results of cosegregation analyses supported pathogenicity of potentially causal mutations in 42 families. In 6 results argued against pathogenicity.
CONCLUSIONS: Gene-panel based NGS results in a substantial increase in diagnostic yield for DCM patients compared to previous results of Sanger sequencing most prevalent genes (55% vs 20-25%). TTN mutations are most prevalent in DCM patients (14%). Higher diagnostic yields are achieved for patients fulfilling DCM and HCM criteria. Cosegregation analyses further support pathogenicity of potentially causal mutations. Together, our gene-panel based approach greatly improved genetic diagnostics in cardiomyopathies.
Article Information
vol. 130 no. Suppl 2 A15882
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Jan D Jongbloed1;
- Anna Pósafalvi1;
- Renée C Niessen1;
- Yvonne M Hoedemaekers1;
- Paul A van der Zwaag1;
- Daniela Q Barge-Schaapveld2;
- Sebastiaan R Piers3;
- Jasper J van der Smagt4;
- Folkert W Asselbergs5;
- Rudolf A de Boer6;
- Maarten P van den Berg6;
- Rowida Almomani1;
- Richard J Sinke1;
- J. P van Tintelen1
- 1Dept of Genetics, Univ Med Cntr Groningen, Univ of Groningen, Groningen, Netherlands
- 2Dept of Clinical Genetics, Leiden Univ Med Cntr, Leiden, Netherlands
- 3Dept of Cardiology, Leiden Univ Med Cntr, Groningen, Netherlands
- 4Dept of Med Genetics, Univ Med Cntr Utrecht, Utrecht, Netherlands
- 5Dept of Cardiology, Univ Med Cntr Utrecht,, Utrecht, Netherlands
- 6Dept of Cardiology, Univ Med Cntr Groningen, Univ of Groningen, Groningen, Netherlands
Abstract 15805: PCSK9 R46L, Lower LDL and Cardiovascular Disease Risk in Familial Hypercholesterolemia
Alexis Baass
Circulation. 2014;130:A15805
Abstract
Introduction: Proprotein convertase subtilisin/kexin type 9 (PCSK9) is a down-regulator of the low density lipoprotein receptor (LDLR). Familial hypercholesterolemia (FH) is a autosomal monogenic disease associated with high LDL-C concentration and cardiovascular risk.
Hypothesis: This study aimed to examine whether the PCSK9 R46L loss of function mutation found in a cohort of FH patients will be associated with lower LDL-C and cardiovascular risk.
Methods: We studied FH patients attending the IRCM Lipid Clinic and whose DNA genotyping was positive for LDLR mutations. The presence of the PCSK9 loss of function R46L missense mutation was determined among a cohort of 582 FH patients by sequencing.
Results: Frequency of the R46L variant was 3%. Comparison of their lipid profile showed that carriers had significantly reduced LDL-C (11%, p<0.001), TC (9%, p<0.01), ApoB (10%, p<0.01) and non-HDL (12%, p<0.001) concentrations compared to non-carriers. The analysis of physical xanthomata among both groups, showed a decreased average number of xanthoma per individual in R46L carriers (0.33 and 0.76 respectively, p<0.001). Importantly, the R46L mutation was associated with a significant 66% (p=0.05) lower risk of cardiovascular events compared to non-carriers.
Conclusion: Our study showed that the presence of the PCSK9 loss of function R46L mutation in the genotype of FH patients is beneficial for the lipid homeostasis and down-regulate the effect of the LDLR mutation in terms of accumulation of LDL-C, ApoB , total cholesterol, non-HDL and thereby, lowers the coronary heart disease risk of PCSK9 LOF mutation carriers. It is therefore very likely that anti-PCSK9 therapy will be useful in reducing cardiovascular risk in FH patients.
Article Information
vol. 130 no. Suppl 2 A15805
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Alexis Baass
- Medicine, McGill Univ, Montreal, Canada
Abstract 15011: Genetic Variants in ABCG5/G8 Are Associated With Primary Hypercholesterolemia
Itziar Lamiquiz-Moneo, Ana M Bea, Rocío Mateo-Gallego, Ana Cenarro, Lucia Baila-Rueda, Fernando Civeira, Isabel De Castro-Orós
Circulation. 2014;130:A15011
Abstract
Introduction: A large proportion of subjects with primary hypercholesterolemia do not have a causative mutation in LDLR, APOB, or PCSK9. GWAs have identified ABCG5 and ABCG8 associated with LDL cholesterol (LDLc) levels. ABCG5/G8 encode for transporters that play an important role in decreasing sterols intestinal absorption and promoting its biliary excretion. The downexpression in ABCG5/G8 produce an increase of cholesterol into the VLDL and LDL leading to hypercholesterolemia.
Hypothesis: ABCG5/G8 genetic variants are associated with primary hypercholesterolemia.
Methods: We have sequenced ABCG5 and ABCG8 promoters, coding regions, and intron-exon boundaries in 191unrelated subjects with LDLc >95th percentile, triglycerides <200 mg/dl and body mass index (BMI) <27.5 kg/m2 and no mutation in PCSK9, LDLR and APOB. Bioinformatical analysis were carried out with MutationTaster, PolyPhen-2 and SIFT and statistical analysis were performed using SPSS software v.20.
Results: We have identified 21 and 25 previously identified variants in ABCG5 and ABCG8 respectively. In addition, we have found a new ABCG5 variant p.(Asn285Ser). By comparing the allelic frequencies of those variants with the reported by 1000 Genomes Project we have found differences with statistical significance in 10 and 7 SNPs in ABCG5 and ABCG8 respectively. Three of these changes have been predicted as damaging or disease causing by Bioinformatical analysis: p.Arg50Cys, p.Asn440Lys and p.Val151Val. Multivariate lineal regression using LDLc as dependent variable has shown that c.502-273A>G in ABCG5, BMI and c.*380T>G in ABCG5 explain independently of age and gender the 13,1% of the LDLc variability (p=0.002).
Conclusions: Common and rare genetic variants in ABCG5/G8 are associated with primary hypercholesterolemia. Common ABCG5 variants explain a substantial proportion of LDLc in primary hypercholesterolemia, suggesting a polygenic background in some cases with this phenotype.
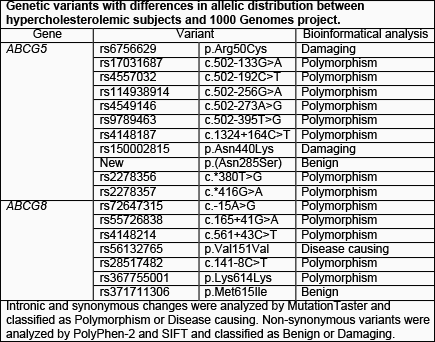
Article Information
vol. 130 no. Suppl 2 A15011
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Itziar Lamiquiz-Moneo;
- Ana M Bea;
- Rocío Mateo-Gallego;
- Ana Cenarro;
- Lucia Baila-Rueda;
- Fernando Civeira;
- Isabel De Castro-Orós
- Lipid Unit and Molecular Rsch Laboratory, Hosp Universitario Miguel Servet, Zaragoza, Spain
Abstract 11449: Influence of Exonic Variation on Heparin-Induced Thrombocytopenia
Jason H Karnes, Joshua C Denny, Robert M Cronin, Christian M Shaffer, Erica A Bowton, James D Cowan, Jonathan D Mosley, Sara L Van Driest, Peter E Weeke, Quinn S Wells, Dan M Roden
Circulation. 2014;130:A11449
Abstract
Introduction: Heparin-induced thrombocytopenia (HIT) is an immune-mediated, potentially fatal adverse effect of heparin treatment. Here we test the hypotheses that exonic variants are associated with HIT and that genes involved in HIT pathophysiology are enriched with rare exonic variation.
Methods: We identified HIT cases and heparin-exposed controls in an electronic medical record coupled to a DNA biobank. Controls were matched to cases based on age, gender, and heparin anticoagulant (unfractionated versus low molecular weight heparin). Genotypes were determined using the HumanExome BeadChip and individual variants were analyzed for association with HIT using multivariable logistic regression in a dominant model. Aggregated rare variant analyses (unidirectional variable threshold [VT] and bidirectional sequence kernel association test [SKAT]) were performed to identify HIT-associated genes and were restricted to amino-acid coding (AAC) variants (missense, non-synonymous, or frameshift) with minor allele frequency less than 0.01.
Results: We identified 77 HIT cases and 345 matched controls. After adjustment for age, gender, heparin anticoagulant, and first two principal components, the variant most strongly associated with HIT was in the human leukocyte antigen class II, DR alpha gene (HLA-DRA,odds ratio 3.81 [2.12-6.83], p=1.01×10-5). While this association was nominally significant, it did not reach a Bonferroni-corrected significance level. In VT analysis, rare ACC variants were most strongly enriched in HIT cases for the genes IFI44L (interferon-induced protein 44-like, p=0.005) and CXCL11 (chemokine [C-X-C motif] ligand 11, p=0.008). IFI44L and CXCL11 were also enriched in HIT cases in SKAT analysis (p=0.0003 and p=0.0001 respectively).
Conclusions: Using single variant and aggregated rare variant analyses, we implicate variants from HLA-DRA and rare variants from IFI44L and CXCL11 as risk factors for HIT. These genes represent biologically plausible candidates, considering the putative role of class II HLA molecules, T cell chemotaxis, and interferons in immune response to heparin.
Article Information
vol. 130 no. Suppl 2 A11449
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Jason H Karnes;
- Joshua C Denny;
- Robert M Cronin;
- Christian M Shaffer;
- Erica A Bowton;
- James D Cowan;
- Jonathan D Mosley;
- Sara L Van Driest;
- Peter E Weeke;
- Quinn S Wells;
- Dan M Roden
- Medicine, Vanderbilt Univ, Nashville, TN
Abstract 20318: Elevated Levels of Lipoprotein(a) Are Causally Associated With Chronic Kidney Disease
Jennie Lin, Wei Zhao, Daniel J Rader, Muredach Reilly, Danish Saleheen
Circulation. 2014;130:A20318
Abstract
Introduction: It remains unknown whether circulating Lipoprotein(a) [Lp(a)] levels and isoform size of apolipoprotein(a) [apo(a)] bound to Lp(a) are causally relevant to CKD.
Methods: We conducted measurements for Lp(a) levels and apo(a) isoform size in 10,765 participants enrolled in the Pakistan Risk of Myocardial Infarction Study (PROMIS). 1,420 participants were diagnosed to have CKD based on estimated glomerular filtration rate (eGFR) less than 60 mL/min/1.73 m2. Lp(a) levels were measured using a high-sensitivity immunoturbidimetric assay, whereas apo(a) isoform size was measured using a validated qPCR assay that assesses CNV repeats in the exon-4 of the LPA gene. We compared the Odds Ratio (OR) for CKD conferred by observed increases in Lp(a) levels with equivalent differences caused by genetic variation due to rs10455872, a variant known to regulate Lp(a) levels. Data on 72,369 participants were available for rs10455872 in association with CKD through combined analyses of PROMIS and the CKDGEN consortium.
Results: Per standard deviation increase in log-Lp(a) levels was associated with increased risk of CKD in PROMIS independent of age, sex, type-2 diabetes, history of hypertension, and tobacco use (OR: 1.08; 95% CI. 1.01 – 1.14), whereas no significant association was observed between apo(a) isoform size and CKD risk. Per copy of the minor allele at rs10455872 was associated with an increase in Lp(a) levels in PROMIS participants (P-value 1.2×10-8). Genetically elevated Lp(a) levels by the minor allele at rs10455872 were associated with increased risk of CKD (OR: 1.02: p-value 3.5×10-3), which was directionally consistent with the OR observed for circulating Lp(a) concentration.
Conclusions: Elevated levels of Lp(a) may have a causal but modest effect on the risk of developing CKD.
Article Information
vol. 130 no. Suppl 2 A20318
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Renal Electrolyte and Hypertension Div, Dept of Medicine, Univ of Pennsylvania, Philadelphia, PA
- 2Translational Medicine and Human Genetics, Univ of Pennsylvania, Philadelphia, PA
- 3Cardiovascular Institute, Univ of Pennsylvania, Philadelphia, PA
- 4Cardiovascular Institute, Dept of Epidemiology, Univ of Pennsylvania, Philadelphia, PA
Abstract 20402: Exome Genotyping of Dallas Heart Study Reveals Putative Cardiac Disease-Causing Mutations: Disconnect Between Genotype and Phenotype
Douglas Stoller, Jonathan Cohen, Helen Hobbs
Circulation. 2014;130:A20402
Abstract
Next generation sequencing has increased the scope and availability of exome sequencing to clinical practice. A major challenge of using the new technology is handling incidental findings. The American Society of Medical Genetics (ACMG) recommends that mutations in 56 genes, including 20 genes that cause heart disease, be reported to patients, regardless of the indication for sequencing. The implications of applying these recommendations to clinical practice are unclear. Here we screened participants of the Dallas Heart Study (n=4928), with the Illumina HumanExome BeadChip array. A total of 46 sequence variants in 6 of the ACMG cardiac genes were identified. Six pathogenic variants were found in 36 participants (0.7% of the cohort). Those with the putative pathologic variants in cardiac genes were not more likely to have a positive family history for cardiac disease or sudden death (69% vs 65%; p=0.81) or an elevated serum cardiac troponin (33% vs 22%; p=0.41] when compared to carriers of benign or low frequency variants of unknown significance (VUS) in the same genes (25 variants in 78 participants). No difference in median (interquartile range) of BNP [7.1 pg/ml (0.7-47 pg/ml) vs 4.9 pg/ml (0-20 pg/ml); p=0.34), ejection fraction [70% (63-76%) vs 71% (69-77%); p=0.41] or LV mass [151 g (115-190 g) vs 143 g (99-176 g); p=0.52] were identified between the two groups who had variants in GLA, MYBPC3, and TNNT2. The QTc interval was similar in those with putative pathogenic KCNQ1 variants and benign/VUS variants [416 ms (406-437 ms) vs 410 ms (401-427ms)]. Our data indicate that cardiac incidental findings will be identified in >1% of the population and that a significant proportion of these individuals will not have clinically significant heart disease.
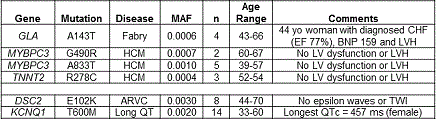
Article Information
vol. 130 no. Suppl 2 A20402
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Douglas Stoller;
- Jonathan Cohen;
- Helen Hobbs
- Dept of Internal Medicine, Univ of Texas Southwestern, Dallas, TX
Abstract 18993: Genetic Variants Primarily Associated With Inflammatory Bowel Disease Do Not Associate With Coronary Artery Disease
Henning Jansen, Wolfgang Lieb, Paola G Ferrario, Nelson P Christopher, Sekar Kathiresan, Reilly P Mudedach, Themistocles L Assimes, Eric Boerwinkle, Alistair S Hall, Christian Hengstenberg, Ruth McPherson, Robert Roberts, Nilesh J Samani, Jeanette Erdmann, Heribert Schunkert
Circulation. 2014;130:A18993
Abstract
Background: Patients suffering from inflammatory bowel disease (IBD) are at an increased risk for coronary artery disease (CAD). Despite the evident coincidence of these inflammatory conditions, a causal role of IBD for CAD has never been proven. Recently, many single nucleotide polymorphisms (SNPs) affecting IBD risk have been identified by genome-wide association studies (GWAS). If IBD acts as a causal trigger for CAD, one might expect that the IBD-associated SNPs alter the risk of CAD.
Methods and Results: We identified a total of 163 SNPs in the literature associated with IBD at a genome-wide significant level of p<5×10-8 and tested these for association with CAD in CARDIoGRAM, a meta-analytical dataset containing GWAS data from 22,233 CAD cases and 64,762 controls. Out of 147 SNPs, which passed quality control in CARDIoGRAM, 99 SNPs affected risk of ulcerative colitis (UC) and Crohn`s disease (CD) with consistent directionality. Approximately equal numbers of these IBD risk alleles produced odds ratios (ORs) for CAD greater (n=40) and smaller (n=59) than OR=1.0, consistent with no association between IBD-SNPs and CAD. Similar results were obtained by analyzing SNPs which only associate with either UC or CD. In total, 20 of the 147 SNPs are associated exclusively with UC and 28 are associated exclusively with CD, but not with UC. Neither UC- nor CD-associated risk alleles showed a positive association with CAD, i.e. the distribution of ORs for CAD above and below 1.0 was similar. Two UC-associated SNPs (rs4722672 and rs17229285) and four CD-associated SNPs (rs6679677, rs10061469, rs2024092, rs6651252) produced p-values below the 5-percent level with inconsistent directionality, none was study-wide significantly associated with CAD.
By contrast, blood pressure- (21 of 25 SNPs) and LDL cholesterol-associated SNPs (18 of 24 SNPs) increased CAD risk consistently to their effects on the underlying risk factors (OR>1, p=0.0008 and p=0.03, respectively).
Conclusion: Analyzing genetic information for the association of IBD and CAD, we found no evidence for a causal role of IBD in the pathogenesis of CAD. Rather, shared non-genetic factors might contribute to the coincidence of IBD and CAD.
Article Information
vol. 130 no. Suppl 2 A18993
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Henning Jansen1;
- Wolfgang Lieb2;
- Paola G Ferrario3;
- Nelson P Christopher4;
- Sekar Kathiresan5;
- Reilly P Mudedach6;
- Themistocles L Assimes7;
- Eric Boerwinkle8;
- Alistair S Hall9;
- Christian Hengstenberg1;
- Ruth McPherson10;
- Robert Roberts10;
- Nilesh J Samani4;
- Jeanette Erdmann3;
- Heribert Schunkert1
- 1Klinik für Erwachsenenkardiologie, Deutsches Herzzentrum München, Deutsches Zentrum für Herz-Kreislauf-Zentrum (DZHK), Munich, Germany
- 2Institut für Epidemiologie, Christian-Albrechts Universität zu Kiel, Kiel, Germany
- 3Institut für integrative und experimentelle Genomik, Universität zu Lübeck, Luebeck, Germany
- 4Dept of Cardiovascular Sciences, Univ of Leicester, Leicester, United Kingdom
- 5Massachusetts General Hosp, Cardiovascular Rsch Cntr and Cardiology Div, Boston, MA
- 6Klinik für Erwachsenenkardiologie, The Cardiovascular Institute, Philadelphia, PA
- 7Dept of Medicine, Stanford Univ Sch of Medicine, Stanford, CA
- 8Human Genetics Cntr and Institute of Molecular Medicine, Univ of Texas Health Science Cntr, Houston, TX
- 9Div of Epidemiology, Leeds Institute of Genetics, Health and Therapeutics, Univ of Leeds, Leeds, United Kingdom
- 10The John & Jennifer Ruddy Canadian Cardiovascular Genetics Cntr, Univ of Ottawa Heart Institute, Ottawa, Canada
Abstract 18169: Genetic Association Study of Circulating Galectin-3 in the General Community Identifies a Variant Associated With Myocardial Infarction
Naveen Pereira, Christopher Scott, Gregory Jenkins, Dennis Robinson, Nirubol Tosakulwong, John Burnett, Richard Weinshilboum, Margaret Redfield
Circulation. 2014;130:A18169
Abstract
Introduction: Galectin (Gal)-3 deficiency has been associated with accelerated atherosclerosis and proinflammatory pathways in mice but its implication in human atherosclerosis is unknown. The variability in Gal-3 levels may be inherited. We sought to determine the genetic variants associated with circulating Gal-3 levels and its impact on vascular events in the general community.
Methods: Serum Gal-3 levels were measured in a random sample of 1,838 Olmsted County, MN residents aged ≥ 45 years. Mean follow up time was 11.1 years. Genotyping was performed by using the Cardio-MetaboChip in 920 random individuals who served as the discovery cohort and replicated in a separate population of 918 subjects. An additive genotype model based on number of copies of the minor allele was used for analyses.
Results: There were 4 genome-wide significant (p≤3.9 X 10-7) single nucleotide polymorphisms (SNPs) in chromosome 14 in GCH1 and FBXO034 genes (rs943912, rs8022453, rs10131232, rs7148669) that were associated with Gal-3 levels in the discovery cohort (Figure). Three of these SNPs, all in GCH1, were replicated after correction for multiple testing. The most significant SNP, rs10131232, was intronic in GCH1 gene with a minor allele frequency=0.342 and was associated with lower Gal-3 levels (β=-0.095, p=1.35X10-17 in the discovery cohort, and β=-0.086, p=4.41X10-17 in the replication cohort). The minor allele of rs10131232 was associated with an increased risk for myocardial infarction (MI) (HR=1.3, p=0.03) as compared to the major allele. Association with MI was maintained after controlling for age, sex, body mass index, diabetes mellitus, hypertension, smoking, and total cholesterol (HR=1.2, p=0.04).
Conclusions: A genetic association study of circulating Gal-3 resulted in the identification and replication of 3 SNPs in the GCH1 gene. The most significant SNP rs10131232 was associated with lower Gal-3 levels and an increased risk for MI in the general community.
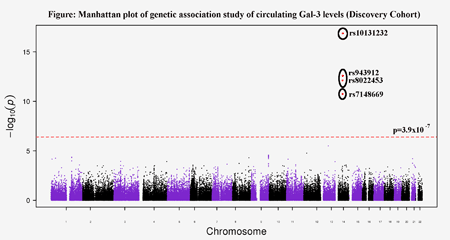
Article Information
vol. 130 no. Suppl 2 A18169
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Naveen Pereira;
- Christopher Scott;
- Gregory Jenkins;
- Dennis Robinson;
- Nirubol Tosakulwong;
- John Burnett;
- Richard Weinshilboum;
- Margaret Redfield
- Medicine, Mayo Clinic, Rochester, MN
Abstract 17297: 9p21 CAD Risk Allele is Protective Against Calcific Aortic Stenosis
Muntaser D Musameh, Peter S Braund, Christopher P Nelson, Jay Gracey, Matthew Denniff, John Thompson, Gerry P McCann, Matthew J Bown, David Sprigings, Nilesh J Samani
Circulation. 2014;130:A17297
Abstract
The 9p21 locus has emerged as one of the strongest risk variants associated with coronary artery disease (CAD). The risk allele for CAD is also associated with other vascular pathologies, including abdominal aortic aneurysms and intracranial aneurysms. Calcific aortic stenosis (AS) and CAD share some risk factors and histopathologic features. However, a considerable proportion of severe AS patients have no significant CAD, indicating the presence of unique mechanisms underlying each condition. To date only one common genetic variant, rs10455872 in the LPA gene, has been robustly associated with risk of AS. Here we investigated the association of the lead CAD-associated variant (rs1333049) at the 9p21 locus with the risk of AS in 1044 cases and 4527 controls. We also typed the LPA SNP (rs10455872) as a validated variant. All cases had either echo confirmed AS of abnormal valve morphology and peak velocity across the aortic valve of more than 2.5 meters/second, or they had previously undergone surgery for severe AS. We used logistic regression analysis to investigate the additive effect of both variants on risk of AS, adjusting for CAD status. For the LPA locus, we observed the previously reported association of the G allele at rs10455872 with risk of AS (OR 1.23; 95% CI, 1.06 to 1.44, P= 0.008). Interestingly for the 9p21 locus, the CAD associated allele (C) for rs1333049 was associated with a lower risk of AS (OR 0.88; 95% CI, 0.80 to 0.98, P= 0.014). In an additional analysis, we examined the association of rs1333049 with AS in our 1044 cases against an independent set of 828 controls with no history of AS, and the results were consistent with the initial findings (OR 0.76; 95% CI, 0.66 to 0.87, P= 0.0001). This unexpected finding requires further validation, but suggests that the 9p21 locus CAD-associated variant is not uniformly deleterious for other vascular pathologies – and for AS may be protective. If the 9p21 finding is confirmed, then understanding the differential effect of this locus on AS and CAD may help to better understand the specific pathogenesis of each condition.
Article Information
vol. 130 no. Suppl 2 A17297
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Muntaser D Musameh1;
- Peter S Braund1;
- Christopher P Nelson1;
- Jay Gracey1;
- Matthew Denniff1;
- John Thompson1;
- Gerry P McCann1;
- Matthew J Bown1;
- David Sprigings2;
- Nilesh J Samani1
- 1Cardiovascular Sciences, Univ of Leicester, Leicester, United Kingdom
- 2Cardiology, Northampton General Hosp NHS Trust, Northampton, United Kingdom
Abstract 16260: Aspirin-Responsive Platelet Genes Are Associated With Platelet Function and Cardiovascular Events
Sunil Suchindran, Tiffany Himmel, Thomas L Ortel, Richard C Becker, William E Kraus, L K Newby, Geoffrey S Ginsburg, Deepak Voora
Circulation. 2014;130:A16260
Abstract
Background: Aspirin prevents myocardial infarction (MI); the pathways with which aspirin interacts are not fully described. We hypothesized that genes that changed in response to aspirin exposure would be associated with platelet function and death/MI.
Methods: Discovery (healthy volunteers; n=53) and Validation (diabetic patients; n=45) cohorts were exposed to aspirin 325mg/day. Before and after aspirin exposure, we measured platelet function by aggregometry and gene/platelet protein expression by whole blood RNA microarray analysis and unbiased proteomics of purified platelet protein, respectively. Gene Set Enrichment Analysis (GSEA) identified gene sets that changed in response to aspirin. Paired t-tests and regression were used to test individual genes and proteins for changes in expression with aspirin or association with platelet function, respectively. Cox regression models tested gene sets in aggregate using the 1st principal component of gene expression for association with death/MI in patients (n =638) using RNA collected at cardiac catheterization.
Results: In the discovery cohort we identified 25 gene sets that changed in response to aspirin (family wise error rate [FWER] < 0.05). Of these, we chose to follow up on one set of 43 genes because it represented abundant platelet genes. This gene set was down regulated in the discovery and validation cohorts (FWER p = 0.01 and 0.04, respectively). We identified 4 of 43 genes in the platelet protein dataset; the expression of 3 changed (p < 0.03) in response to aspirin and was associated (p < 0.01) with platelet function after accounting for aspirin. The aggregate expression of this platelet gene set was associated with death/MI (p = 0.03) with the majority of genes associated with increased risk. Analysis of individual genes in this gene set identified several (HLA-E, H3F3A, GNAS, FTH1, TUBA4A, OAZ1, FLNA) that were significantly (p < 0.05) down-regulated by aspirin and also associated with an increased risk of death/MI.
Conclusion: By using aspirin as a probe to uncover novel biology we identified a set of aspirin-responsive platelet genes associated with an increased risk of cardiovascular events. These findings may help to explain the effects of aspirin on preventing MI.
Article Information
vol. 130 no. Suppl 2 A16260
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Sunil Suchindran1;
- Tiffany Himmel1;
- Thomas L Ortel1;
- Richard C Becker2;
- William E Kraus1;
- L K Newby3;
- Geoffrey S Ginsburg1;
- Deepak Voora1
- 1Medicine, Duke Univ, Durham, NC
- 2Medicine, Univ of Cincinnati College of Medicine, Cincinnati, OH
- 3Medicine, Duke Clinical Rsch Institute, Durham, NC
Abstract 15963: Toward Development of a Genetic Risk Score for Sudden Cardiac Death
Adriana Huertas-Vazquez, Christopher P Nelson, Janet S Sinsheimer, Kyndaron Reinier, Audrey Uy-Evanado, Carmen Teodorescu, Jo Ayala, Karen Gunson, Janathan Jiu, Peter S Braund, Panos Deloukas, Alistair S Hall, Anthony J Balmforth, Nilesh J Samani, Sumeet S Chugh
Circulation. 2014;130:A15963
Abstract
Introduction: Genome-wide association studies have identified multiple common variants associated with risk of SCD. However, independently these loci make modest contributions to disease risk. The objective of this study was to develop a genetic risk score (GRS) for SCD to evaluate the cumulative effects of these variants on SCD risk.
Methods: We identified variants with established associations to SCD (14 SNPs). To these we added SNPs associated with intermediate phenotypes of SCD including those involved in cardiac ventricular conduction (QRS duration, 26 SNPs), repolarization (QT interval, 9 SNPs) and heart rate (18 SNPs). After quality control, a total of 67 SNPs were investigated. The GRS was calculated using the weighted approach based on the number of risk alleles weighted by the beta coefficients derived from the original studies. GRSs were tested for association using logistic regression models. A total of 966 cases from an ongoing prospective community-based evaluation of SCD in a large U.S. community and 1,926 subjects with coronary artery disease (CAD) from the Wellcome Trust Case Control Consortium (WTCCC+) were included in this study.
Results: The highest increased risk of SCD was observed with the combination of two previously associated SCD SNPs, rs3010396 within CASQ2 and rs6730157 within RAB3GAP1 (OR= 2.43 [1.8-3.2], P=7.41×10-10). CASQ2 and RAB3GAP1 have been previously implicated in calcium signaling and heart disease. Modest associations were found for a GRS composed of 14 SCD SNPs (OR=1.17 [1.05-1.29], P= 0.002). The results remained significant after adjusting for multiple testing. We did not observe significant associations for SNPs linked to the other investigated traits, for individual loci or the overall combined risk score.
Conclusions: These findings suggest that the cumulative effects of SCD-associated gene variants may contribute to improved SCD prediction. Additional studies of cumulative genetic risk in larger, well-phenotyped populations are warranted.
Article Information
vol. 130 no. Suppl 2 A15963
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Adriana Huertas-Vazquez1;
- Christopher P Nelson2;
- Janet S Sinsheimer3;
- Kyndaron Reinier1;
- Audrey Uy-Evanado1;
- Carmen Teodorescu1;
- Jo Ayala1;
- Karen Gunson4;
- Janathan Jiu5;
- Peter S Braund6;
- Panos Deloukas7;
- Alistair S Hall8;
- Anthony J Balmforth9;
- Nilesh J Samani6;
- Sumeet S Chugh1
- 1Heart Institute, Cedars-Sinai Med Cntr, Los Angeles, CA
- 2Dept of Cardiovascular Sciences, Univ of Leicester, Leicester, United Kingdom
- 3Biomathematics and Human Genetics, Univ of California, Los Angeles, Los Angeles, CA
- 4karen.gunson@state.or.us, Oregon Health and Science Univ, Portland, OR
- 5Emergency Medicine, Oregon Health and Science Univ, Portland, OR
- 6Cardiovascular Sciences, Univ of Leicester, Leicester, United Kingdom
- 7Human Genetics, Wellcome Trust Sanger Institute, Hinxton, United Kingdom
- 8Clinical Cardiology and Epidemiology, Univ of Leeds, Leeds, United Kingdom
- 9Clinical Cardiology, Univ of Leeds, Leeds, United Kingdom
Abstract 12582: Genetically Reduced High-Density Lipoprotein Cholesterol and Risk of Type 2 Diabetes: A Mendelian Randomization Study
Christiane L Haase, Anne Tybjaerg-Hansen, Borge G Nordestgaard, Ruth Frikke-Schmidt
Circulation. 2014;130:A12582
Abstract
Introduction: Epidemiologically low levels of high-density lipoprotein (HDL) cholesterol consistently associate with increased risk of type 2 diabetes. The causal nature of this association is unclear.
Hypothesis: Using Mendelian randomization, we tested whether low levels of HDL cholesterol causally influence type 2 diabetes.
Methods: In a prospective study of 47,627 individuals from the general population, we tested whether low HDL cholesterol predicted risk of type 2 diabetes. Using a combined genotype score or allele numbers of five HDL genes, we tested whether genetic variants associated with low HDL cholesterol levels also associated with an increased risk of type 2 diabetes.
Results: Risk of type 2 diabetes increased with decreasing levels of HDL cholesterol (P for trend=3×10-58). HDL cholesterol decreasing gene scores and allele numbers associated with up to -13% and -20% decreases in HDL cholesterol levels. The corresponding theoretically predicted hazard ratios (HRs) for type 2 diabetes were 1.44 (1.38-1.52) and 1.77 (1.61-1.95), while the genetic estimates were non-significant (P for trend: 0.41 and 0.55). Genetic risk ratios for a 50% reduction in HDL cholesterol were 0.66 (0.33-1.33) and 0.74 (0.37-1.47) for HDL cholesterol decreasing gene scores and allele numbers, respectively, compared to the corresponding observational HR=3.81 (3.30-4.39) (P for comparison: 2×10-6 and 6×10-6, respectively).
Conclusions: Genetically reduced HDL cholesterol does not associate with an increased risk of type 2 diabetes, suggesting that the level of HDL cholesterol is not a causal risk factor for type 2 diabetes.
Article Information
vol. 130 no. Suppl 2 A12582
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Dept. Clin. Biochem., Rigshospitalet, Copenhagen Univ Hosp, Copenhagen, Denmark
- 2Dept. Clin. Biochem., Herlev Hosp, Copenhagen Univ Hosp, Copenhagen, Denmark
Abstract 11864: Examining Rare and Low-Frequency Genetic Variants Previously Associated With Lone or Familial Forms of Atrial Fibrillation in an Electronic Medical Record System: A Cautionary Note
Peter Weeke, Joshua C Denny, Lisa Basterache, Christian Shaffer, Erica Bowton, Christiana Ingram, Dawood Darbar, Dan M Roden
Circulation. 2014;130:A11864
Abstract
Introduction: Studies in individuals or small kindreds have implicated rare variants in 25 different genes in lone and familial atrial fibrillation (AF) using linkage and segregation analysis, functional characterization, and rarity in public databases. Here we used a cohort of 20,204 patients of European ancestry (EA) or African ancestry (AA) (n=18,424 and n=1,780, respectively) with electronic medical records (EMRs) and exome chip data to compare the frequency of AF among carriers and non-carriers of these rare variants.
Methods and Results: The exome chip included 19/115 rare variants, in 9 genes, previously associated with lone or familial AF. Using validated algorithms querying a combination of clinical notes, structured billing codes, ECG reports, and procedure codes, we identified 1,056 AF cases (>18 years) and 19,148 non-AF controls (>50 years) with available genotype data on the Illumina HumanExome BeadChip v.1.0 in the Vanderbilt EMR-linked DNA repository, BioVU. While known correlations between AF and common variants at 4q25 were replicated, none of the 19 variants previously associated with AF were overrepresented among AF cases (P >0.1 for all). The frequency of variant carriers among non-AF controls was >0.1% for 14/19 polymorphisms; 9 of 19 polymorphisms were absent in EA AF cases, but were present in 1-35 (0.01-0.2%) non-AF EA controls. The greatest numbers of risk allele carriers among EA AF cases were identified for the following variants: Phe2004Leu in SCN5A (13 [1.3%] AF cases vs. 179 [1.0%] non-AF controls, p=0.42), Gln76Glu in GREM2 (10 [1.0%] vs. 133 [0.8%], p=0.76), Ser64Arg in NPPA (5 [0.5%] vs. 120 [0.7%], p=0.69), and Arg1193Gln in SCN5A (4 [0.4%] vs. 33 [0.2%], p=0.14). Repeat analyses using EA non-AF controls aged >60 (n=14,904) >70 (n=9,670), and >80 (n=4,729) years old did not influence these findings. Analysis among AA yielded similar results.
Conclusion: Rare variants previously implicated in lone or familial forms AF present on the exome chip are detected at low frequencies in a general population but are not associated with AF. These findings emphasize the need for caution when ascribing variants as pathogenic or causative.
Article Information
vol. 130 no. Suppl 2 A11864
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Peter Weeke;
- Joshua C Denny;
- Lisa Basterache;
- Christian Shaffer;
- Erica Bowton;
- Christiana Ingram;
- Dawood Darbar;
- Dan M Roden
- Medicine, Vanderbilt Univ, Nashville, TN
Abstract 20276: Tetralogy of Fallot With Hypoplastic Branch Pulmonary Arteries: Aggressive Patch Augmentation Improves Short-Term Geometry but Increases Risk of Late Re-Interventions
Travis J Wilder, Glenn Van Arsdell, Eric Pham-Hung, Michael Gritti, Saro Hussain, Christopher A Caldarone, Andrew Redington, Edward J Hickey
Circulation. 2014;130:A20276
Abstract
Introduction: When encountering hypoplastic branch pulmonary arteries (PA) during tetralogy of Fallot repair (TOF), strategies include: 1) patch augment to hilum (PATCH), 2) extending patches into proximal part only (EXTEND), or 3) leave native vessels uninstrumented in anticipation of growth (NATIVE). We tested outcomes for these opposing strategies.
Methods: We studied all 434 TOF repairs (2000-12, excluding pulmonary atresia). Risk-adjusted models analyzed competing endstates of branch PA reintervention or death. PA growth was explored via repeated measures analysis of 2123 echo measurements. Subgroup analysis of children with branch PA < 4 mm at time of repair was performed.
Results: Overall survival was excellent (99%; 3 deaths). Mean freedom from catheter or surgical re-intervention to branch PAs at 10 years was 84%. In models risk-adjusted for baseline features (including PA size), PATCH augmentation of branch PAs was associated with significantly higher rates of re-intervention (75% freedom; p<.01) versus EXTEND (82%) or NATIVE (86%). Additional risk factors included small indexed branch PAs and transcatheter interventions prior to index TOF repair. Additionally, time-related branch PA growth following full PATCH was significantly less than other children (N=2123 echos; p<.01).
Branch PA < 4mm:
In PATCH(28), EXTEND(60) and REPAIR(75) groups, freedom from re-intervention adjusted for BPA z-score was 60%, 70% and 80% respectively, and patient characteristics were similar (figure). More PATCH children had received PA stents (P=.04), but the majority of all groups had not (figure). Aggressive PATCH strategy was associated with decreased time-related branch PA growth (p=.02, 667 echos).
Conclusions: Aggressive patch augmentation of branch PAs improves short-term geometry but may lead to late stenosis and higher rates of re-intervention. Hypoplastic branch PAs in TOF tend to grow well in their native state or with minimal surgical manipulation.
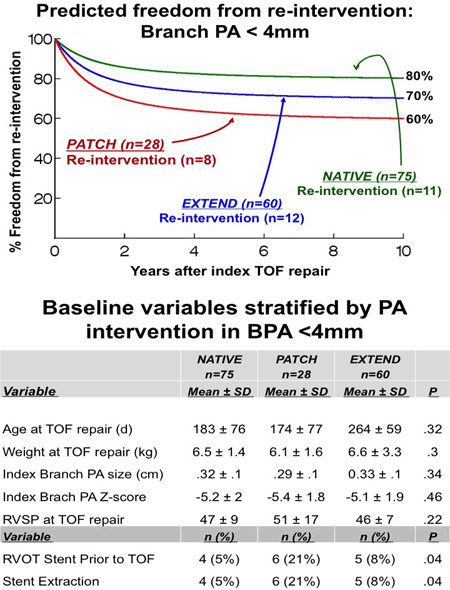
Article Information
vol. 130 no. Suppl 2 A20276
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Travis J Wilder1;
- Glenn Van Arsdell2;
- Eric Pham-Hung3;
- Michael Gritti2;
- Saro Hussain3;
- Christopher A Caldarone3;
- Andrew Redington4;
- Edward J Hickey4
- 1CHSS Data Cntr, The Hosp For Sick Children, Toronto, Canada
- 2The Hosp for Sick Children, The Hosp For Sick Children, Toronto, Canada
- 3Dept of Cardiovascular Surgery, The Hosp For Sick Children, Toronto, Canada
- 4Div of Cardiovascular Surgery, The Hosp For Sick Children, Toronto, Canada
Abstract 18150: Prediction of Imminent Deterioration of Children after Stage I Palliation Using Real-Time Processing of Physiological Data
Craig G Rusin, Sebastian I Acosta, Eric L Vu, Risa B Myers, Kenneth M Brady, Daniel J Penny
Circulation. 2014;130:A18150
Abstract
Patients after stage 1 palliation (S1P) for hypoplastic left heart syndrome (HLHS) and related lesions are at risk of life threatening deterioration resulting in shock, cardiac arrest, & hypoxemia. We hypothesize that these sudden deteriorations may be forecast by subtle, previously unidentified changes in cardiorespiratory dynamics. Identification of these precursors may provide an opportunity for early, life-saving intervention.
We created complete high-resolution physiological recordings for all patients who had a primary admission of S1P after Jan. 1, 2013. We used the SickbayTM system (Medical Informatics Corp, Houston, TX) to collect high frequency physiological waveforms including EKG, ABP, LAP, SpO2 and Chest Impedance (60Hz – 240Hz), as well as HR, RR, Temp. and ST segment vital signs (0.5 Hz) during the patient’s interstage hospitalization. A logistic regression model was constructed to discriminate between physiological characteristics observed in the hours prior to deterioration from the characteristics observed >24 hours prior to or >96 hours after a clinical deterioration. Model validation was done using a standard bagging approach with a REPtree classifier and 10 fold cross validation.
Twenty five patients were included in the study. Of these, 15 (60%) were found to have one or more deterioration events (arrest, CPR, unplanned intubation), with 24 total events observed during the interstage period. Characteristics associated with imminent deterioration were low SpO2 and depressed ST segment. Changes in physiological dynamics could be detected 1-2 hours before overt deterioration occurs (ROC area = 0.89) (Figure 1). This altered physiological state remains for ~96 hours after deterioration.
In conclusion, it is possible to identify clinical deterioration in HLHS patients during their interstage period ~1-2 hours before overt deterioration occurs, providing an opportunity for early, life-saving intervention to be administered.
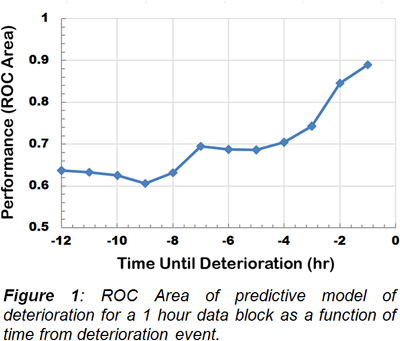
Article Information
vol. 130 no. Suppl 2 A18150
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Pediatric – Cardiology, Baylor College of Medicine, Houston, TX
- 2Pediatric – Anesthesiology, Baylor College of Medicine, Houston, TX
- 3Computer Science, Rice Univ, Houston, TX
Abstract 15372: The Right Ventricular Infundibular Sparing Approach for the Repair of Tetralogy of Fallot Results in Better Intermediate-Term Right Ventricular and Left Ventricular Systolic Function as Assessed by Cardiac MRI
Mary K Olive, Shelby Kutty, Charles D Fraser, Emmett D McKenzie, James M Hammel, Rajesh Krishnamurthy, Shiraz A Maskatia
Circulation. 2014;130:A15372
Abstract
Introduction: The right ventricular infundibular sparing approach (RVIS) to repair Tetralogy of Fallot (TOF) avoids the ventricular incision used in the transventricular (TV) approach.
Hypothesis: We hypothesize that patients repaired with the RVIS approach have less right ventricular dilation and better RV systolic function assessed by cardiac MRI (CMR) than patients who had a TV repair.
Methods: This is a retrospective cohort study of patients who underwent RVIS repair of TOF at one institution or TV repair at a seperate institution and were later evaluated by CMR. Patients were placed into 1 of 4 age-matched groups. We compared right ventricular end diastolic and systolic volumes indexed to body surface area (RVEDVi and RVESVi) and right ventricular ejection fraction (RVEF) as primary endpoints. Secondary endpoints included pulmonary regurgitant fraction (PRF), left ventricular end diastolic volume indexed to body surface area (LVEDVi), and left ventricular ejection fraction (LVEF). Chi-Square, Student’s t, and Mann-Whitney tests were used as appropriate.
Results: Ninety patients were included in the analysis; 45 underwent RVIS repair at median age of 9.8 months (IQR: 6.3-14.6) and 45 underwent TV repair at median age of 4.0 months (IQR: 2.8-6.3), (p<0.01). Heterogeneity existed in the methods used to relieve RV outflow tract obstruction, as efforts were made to spare the pulmonary valve in both groups. None of the patients in the TV group had an initial palliation with a systemic to pulmonary arterial shunt compared to 16 (36%) in the RVIS group (p<0.01). The median age at MRI was 9.3 years (IQR: 6.3-14.1) in the RVIS group and 9.0 years (IQR: 5.8-12.7) in the TV repair group (p=0.3). There were no differences in RVEDVi (122±25 cc/m2 vs 119±32 cc/m2, p= 0.59) or PRF (40±13 vs 37±18, p=0.29) between the RVIS and TV repair groups. Compared to the TV repair group, the RVIS group had higher RVEF (54±6 vs 44±9, p<0.01), lower RVESVi (57±17 cc/m2 vs 67±25 cc/m2, p=0.03), and higher LVEF (61±11 vs 54±8, p<0.01).
Conclusions: Patients who underwent RVIS repair for TOF appeared to have better right and left ventricular systolic function compared to those who had a TV repair. These observations have important implications for prognosis and merit further study.
Article Information
vol. 130 no. Suppl 2 A15372
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Mary K Olive1;
- Shelby Kutty2;
- Charles D Fraser3;
- Emmett D McKenzie3;
- James M Hammel4;
- Rajesh Krishnamurthy5;
- Shiraz A Maskatia6
- 1Section of Pediatric Cardiology, Dept of Pediatrics, Baylor College of Medicine, Houston, TX
- 2Div of Cardiology, Dept of Pediatrics, Univ of Nebraska Med Cntr, Omaha, NE
- 3Div of Congenital Heart Surgery, Dept of Surgery, Baylor College of Medicine, Houston, TX
- 4Div of Cardiothoracic Surgery, Dept of Surgery, Univ of Nebraska Med Cntr, Omaha, NE
- 5Dept of Radiology, Baylor College of Medicine, Houston, TX
- 6Section of Pediatric Cardiology, Baylor College of Medicine, Houston, TX
Abstract 12605: Center Variability in Timing of Stage 2 Palliation and Association With Interstage Mortality: A Report From the National Pediatric Cardiology Quality Improvement Collaborative
Garick Hill, Nancy Rudd, Nancy Ghanayem, David Hehir, Peter Bartz
Circulation. 2014;130:A12605
Abstract
Introduction: The interstage period from discharge following stage 1 palliation (S1P) until stage 2 palliation (S2P) remains high risk. Significant variability between institutions exists around the timing of S2P. We sought to describe the variability in a multi-institution cohort and assess its association with interstage mortality.
Methods: The National Pediatric Cardiology Quality Improvement Collaborative registry, with data from 52 centers, was queried. Patients undergoing a hybrid S1P, transplanted prior to S2P, lost to follow up prior to S2P or deemed not candidates for S2P were excluded. Only centers with 10 or greater patients meeting eligibility were included to reduce the impact of outliers. Centers were divided based on median age at S2P into early (n=15) and late (n=16) centers using a cutoff of 153 days. Groups were compared using Chi-squared or Wilcoxon rank sum test.
Results: The final cohort included 789 patients from 31 centers. Center specific median age at S2P varied from 109 to 214 days, with a center mean of 158 ± 27 days. At S1P, the late centers had a higher prevalence of preoperative ventilation (34.7% vs. 26.9%, p=0.02) and longer average post-S1P duration of intubation (14.4 ± 19.7 vs. 10.2 ± 11.4 days, p<0.001) and S1P hospital length of stay (48.5 ± 30.4 vs. 38.5 ± 22.3 days, p<0.0001). Interstage mortality was significantly higher in centers performing late vs. early S2P (9.9% vs. 5.7%, p=0.03). Interstage event rate (late: 8.2 vs. early: 5.8 deaths per 10000 interstage days) was not different by group (p=0.26), but interstage duration was significantly longer (133.9 ± 71.5 vs. 103.4 ± 37.8 days, p<0.0001) in the late group. Survival to hospital discharge (98.9% in both groups, p>0.98) and hospital length of stay following S2P (late: 15.6 ± 22.3 vs. early: 13.7 ± 22.4, p=0.68) were similar between groups.
Conclusions: In a large multi-institution collaborative, the median age at S2P varies between centers. Centers performing S2P at a later median age have higher interstage mortality. This may be in part due to a higher severity of illness, reflected by higher S1P morbidity in this group. Although optimal timing of S2P remains unclear, centers performing early S2P did not experience worse S2P outcomes, and experienced less interstage mortality.
Article Information
vol. 130 no. Suppl 2 A12605
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Pediatric Cardiology, Children’s Hosp of Wisconsin, Milwaukee, WI
- 2Pediatric Critical Care, Children’s Hosp of Wisconsin, Milwaukee, WI
Abstract 11125: Is the Rastelli Procedure Falsely Maligned?
Mohammed Al-Jughiman, Maryam Al-Omair, Osami Honjo, Erwin Oechslin, Christopher Caldarone, Glen Van Arsdell
Circulation. 2014;130:A11125
Abstract
Background: Surgical options for transposition left ventricular outflow tract obstruction (TGA/LVOTO) are multiple. We and others have noted the Rastelli has high late mortality/morbidity for TGA/VSD/LVOTO. This led to a change in strategy in 2000.
Objective: We hypothesized that patients undergoing a Nikaidoh would have superior outcomes to the Rastelli procedure.
Methods: From ’82-‘13, 157 patients with TGA/LVOTO were operated. Of which 133 (84.7%) had associated VSD. Procedures were: Rastelli (n=41), Mustard ± LVOTO relief (n=41), ASO ± LVOTO relief (n=36), single V (n=26), REV (n=7), and Nikaidoh (n=6). Stratification by era was performed: era 1, ‘82-‘89 (n=70); era 2, ‘90-‘99 (n=50); era 3 ‘00-‘13 (n=37).
Results: Survival at 10 years was not different by strategy: Rastelli 77.9%, Mustard 82.3%, ASO 85.5%, single V 96.2%, REV 100%, and Nikaidoh 83.3% , P=0.24. Survival after the Rastelli improved over eras (at 9 years: era 1=45%, era 2=93%, and era 3=100%, P=0.01) (fig 1A). At 23 years, it was 45% for era 1 and 93% for era 2. Late mortality was independently associated with arrhythmia (OR 3.50; 95% CI 1.15-10.67, P=0.03) but not LVOT gradient (OR 0.95; 95% CI 0.89-1.01, P=0.13) though the first era had a positive time relationship (P=0.01) to increasing gradient (slope 0.49, SE=0.19, 95% CI 0.11-0.88) (LVOT reop fig 1B). The Rastelli and REV were independently associated with all reoperation (OR 10.58; 95% CI 4.23-26.46, P<0.001) and (OR 8.33; 95% CI 1.60-43.17, P=0.01) respectively. The Nikaidoh, Rastelli, and REV had similar RVOT reoperation.
Conclusion: The Rastelli since 1990, had a different outcome than a generation ago potentially suggesting a difference in starting physiologic substrate and or the operation. Greater sample size and follow up will be required to determine if long term outcome differs between the modern Rastelli and Nikaidoh. Given the significant improvement in survival over eras, the current Rastelli remains a viable strategy and is not inferior.
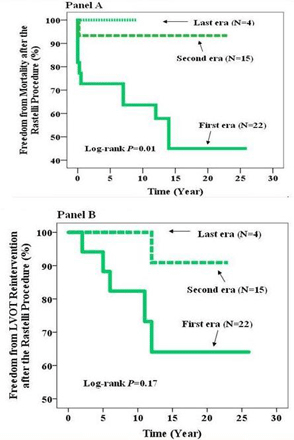
Article Information
vol. 130 no. Suppl 2 A11125
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Mohammed Al-Jughiman1;
- Maryam Al-Omair2;
- Osami Honjo2;
- Erwin Oechslin3;
- Christopher Caldarone2;
- Glen Van Arsdell2
- 1Cardiac Surgery, Univ of Toronto, Toronto, Canada
- 2Cardiac Surgery, Hosp for Sick Children, Toronto, Canada
- 3Peter Munk Cardiac Cntr, Univ Health Network, Toronto General Hosp, Toronto, Canada
Abstract 20164: Profiling of Circulating MicroRNAs After Heart Transplantation and During Acute Cellular and Antibody-Mediated Rejection
Raymond Givens, Danielle Templeton, Ruiping Ji, Peter Kennel, Donna Mancini, Yoshifumi Naka, Hiroo Takayama, Isaac George, P. C Schulze
Circulation. 2014;130:A20164
Abstract
Introduction: Heart transplantation (HTx) is the definitive treatment for advanced heart failure. Despite universal immunosuppressive therapy, the threat of allograft rejection is persistent. Rejection is generally diagnosed by histology of graft myocardium obtained by biopsy which is invasive and prone to sampling error. Study of novel biomarkers such as circulating microRNAs (miRs) may allow noninvasive diagnosis of rejection.
Methods: We studied serum levels of miRs associated with immune cell function (miR-155, 125b, 142-3p, 144, 223-3p, 27a and 101), myocardial damage (miR-1) and immunosuppression (let-7a). Blood was obtained from HTx recipients >2 months post-HTx. Rejection was defined by histology of cardiac biopsy specimens and graded according to ISHLT guidelines. MiRs were analyzed with RT-PCR and normalized to 5S rRNA.
Results: We analyzed samples from 12 healthy volunteers (44±6 yrs), 12 HTx recipients without evidence of rejection (54±11 yrs), 11 patients with acute cellular rejection (ACR; 48±12 yrs), and 6 patients with antibody-mediated rejection (AMR; defined by C4D+ immunohistochemistry; 48±13 yrs). The ACR group included 7 patients with ISHLT 1R/1B rejection and 4 with 2R/3A. 6 of 11 ACR and 5 of 6 AMR patients had evidence of donor-specific antibodies. Let-7a levels were significantly lower among the HTx group vs. controls (0.09±0.13 vs. 1.00±1.53 RU; p=0.02) and increased only among the ACR group (0.44±0.64, p=0.03) vs. the HTx group. MiR-223-3p was lower among HTx (0.31±0.38), ACR (0.33±0.37) and AMR (0.38±0.37) vs. control (1.00±0.87, p<0.01). MiR-101 levels were increased among ACR (1.83±1.61, p<0.01) and AMR groups (1.66±1.24, p<0.01) vs. HTx (0.41±0.51). There were trends towards higher miR-142-3p among ACR (p=0.06) and AMR (p=0.09) vs. the HTx group. Combination of let-7a and miR-101 and 144 identified patients with acute rejection compared to stable HTx patients (ROC AUC 0.82).
Conclusion: Circulating miRNAs related to immune function may aid in diagnosis of cardiac allograft rejection. Our data show that miR-101 and 223-3p and let-7a have diagnostic potential for identifying patients with ACR and AMR. Our ongoing studies will analyze roles for miRs as ancillary markers of graft rejection following HTx.
Article Information
vol. 130 no. Suppl 2 A20164
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Raymond Givens;
- Danielle Templeton;
- Ruiping Ji;
- Peter Kennel;
- Donna Mancini;
- Yoshifumi Naka;
- Hiroo Takayama;
- Isaac George;
- P. C Schulze
- Medicine, Columbia Univ, New York, NY
Abstract 18159: Transcriptomic Profiling of Isogenic PBMC-derived and iPSC-derived Macrophages and Their Polarization
Hanrui Zhang, Chenyi Xue, Rhia Shah, Kate Bermingham, Christine C Hinkle, Wenli Yang, Sager J Gosai, Daniel VanDorn, Stella T Chou, Brian D Gregory, Edward E Morrisey, Mingyao Li, Daniel J Rader, Muredach P Reilly
Circulation. 2014;130:A18159
Abstract
iPSC technology has demonstrated enormous potential for in vitro disease modeling. However, an important issue in the field is to what extent iPSC-differentiated cells adopt the transcriptomic characteristics of the primary somatic cells. To address this question, we performed deep RNA-seq in PBMC-derived iPSC-differentiated macrophages (IPSDM, >95% purity, n=4) and their isogenic human PBMC-derived macrophages (HMDM, by M-CSF) counterparts that were validated to be phenotypically and functionally highly comparable. Multidimensional scaling and hierarchical clustering revealed marked transcriptome changes during iPSC transition to IPSDM, which led to 100 to >4,000-fold decreases in the expression of key pluripotency genes and a concomitant increase in that of key macrophage functional genes with an over-representation of GO terms associated with immune response, defense response, and inflammatory response. Compared with isogenic HMDM, IPSDM revealed ~90% genes to be similarly expressed, but 1,632 genes (~10% of detectable genes at 1stpercentile FPKM) to be differentially expressed (DE) (FDR-adjusted P<0.01, fold-change>2). Genes expressed at higher levels in IPSDM include typical fibroblast markers and genes encoding collagen and extracellular matrix, but not lineage markers of other hematopoietic cells. M1-polarized (by LPS+IFN) cells showed distinct profile from non-polarized and M2-polarized (by IL-4) cells that were relatively less separated. There were marked overlap (>70%) of DE genes between non-polarized and M1-polarized cells in HMDM and IPSDM. Many of the genes expressed at lower levels in non-polarized IPSDM compared with HMDM were upregulated during subsequent polarization to levels comparable to that of M1- or M2- HMDM suggesting that polarization leads to greater convergence of ISPDM and HMDM transcriptome. In summary, at the transcriptome level, IPSDM and HMDM were similar with marked differences between IPSDM and their iPSC precursors. M1 polarization was associated with a dramatic and similar change in the transcriptome of IPSDM and HMDM.
Article Information
vol. 130 no. Suppl 2 A18159
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Hanrui Zhang1;
- Chenyi Xue1;
- Rhia Shah1;
- Kate Bermingham1;
- Christine C Hinkle1;
- Wenli Yang2;
- Sager J Gosai3;
- Daniel VanDorn4;
- Stella T Chou4;
- Brian D Gregory3;
- Edward E Morrisey5;
- Mingyao Li6;
- Daniel J Rader7;
- Muredach P Reilly1
- 1Cardiovascular Institute, Univ of Pennsylvania, Philadelphia, PA
- 2Institute for Regenerative Medicine, Univ of Pennsylvania, Philadelphia, PA
- 3Biology, Univ of Pennsylvania, Philadelphia, PA
- 4Pediatrics, The Children’s Hosp of Philadelphia, Philadelphia, PA
- 5Cell and Developmental Biology, Univ of Pennsylvania, Philadelphia, PA
- 6Biostatistics and Epidemiology, Univ of Pennsylvania, Philadelphia, PA
- 7Institute for Translational Medicine and Therapeutics, Univ of Pennsylvania, Philadelphia, PA
Abstract 17711: Myocardial Long Non-coding RNA Expression Exhibits Chamber- and Disease-Specific Signatures in Human Right Ventricle
Kai-Chien Yang, Thomas G Di Salvo
Circulation. 2014;130:A17711
Abstract
Background: Eukaryotic genome transcription is pervasive; >90% of the mammalian genome is transcribed as long non-coding RNAs (lncRNAs). It is known that human cardiac lncRNA expression is dynamically regulated in the left ventricle (LV) with heart failure (HF). However, it remains unclear how lncRNA expression is regulated in normal and failing right ventricle (RV).
Methods & Results: Paired-end RNA sequencing (RNASeq) was conducted using RNA isolated from paired RV and LV samples from non-failing (NF) hearts (n=5), as well as from the RVs of dilated non-ischemic (DCM, n=11) and ischemic (ICM, n=11) human failing hearts. A total of 970568044 read pairs were obtained. RNASeq analysis revealed high abundance of lncRNAs of mitochondrial origin in both RV (58.8%) and LV (56.5%). Among the 4129 lncRNAs significantly expressed (≥1 reads per killobase exon per million mapped reads) in human myocardium, 439 (10.6%) were differentially expressed (absolute fold-change≧1.2, adjusted P<0.05) between non-failing RV and LV. Hierarchical clustering analyses showed that the expression profiles of lncRNAs, but not mRNAs, distinguished NF RV from NF LV (Figure). There were 667 (459 up,208 down) and 690 (513 up,177 down) lncRNAs dysregulated in the RVs with ICM and DCM, respectively, compared with NF RV samples. Principal component analyses showed that the expression signature of RV lncRNAs, but not mRNAs, discriminates both non-failing from failing RVs and the etiology of heart failure (ICM vs DCM) (Figure).
Conclusion: Human cardiac lncRNA expression exhibits chamber-specific signatures and discriminates RV from LV. RV lncRNAs are dynamically regulated with advanced HF, and the expression profiles of RV lncRNAs discriminate failing hearts of different etiologies. Taken together, these results suggest that lncRNAs may play an important role in shaping chamber-specific ventricular function and may contribute to etiology-specific RV remodeling processes during HF.
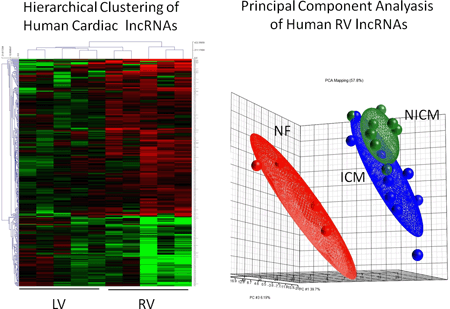
Article Information
vol. 130 no. Suppl 2 A17711
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Dept of Pharmacology, National Taiwan Univ, Taipei, Taiwan
- 2Advanced Heart Failure Program, Vanderbilt Heart and Vascular Institute, Vanderbilt Univ Sch of Medicine, Nashville, TN
Abstract 17732: Identification of Stromal Interaction Molecule 1 Complexes in Cardiac Hypertrophy
Ahyoung Lee, Changwon Kho, Ludovic Bénard, Antoine Chaanine, Roger Hajjar, Jean-Sébastien Hulot
Circulation. 2014;130:A17732
Abstract
Abnormalities in intracellular calcium signaling initiate and promote sustained pathological cardiac hypertrophy. Stromal interaction molecule 1 (STIM1) is an essential calcium sensor that activates calcium influx in response to calcium depletion in the endoplasmic reticulum (ER) in many cells. Previously, we found that STIM1 is activated and functional in hypertrophic cardiomyocytes where it controls store-dependent and -independent calcium influxes that further promote the development of cardiac hypertrophy. To understand the regulatory mechanisms of STIM1 during hypertrophic remodeling, we performed interactome analyses on a rat model of compensated cardiac hypertrophy resulting from pressure overload by thoracic aortic banding. STIM1 complexes were immunoprecipitated from cardiomyocytes isolated from either control or aortic-banded hearts. Electrospray ionization tandem mass spectrometry analysis identified a total of 93 proteins that were reliably enriched in the STIM1 immunoprecipitates in multiple independent experiments. Silver staining of the isolated complexes revealed distinct patterns of associated proteins in the disease states. STIM1 complexes were enriched in hypertrophic hearts with a total of 78 identified proteins as compared to 53 proteins in control hearts. Only 38 proteins were associated with STIM1 in both control and diseased conditions. Next, we performed a functional classification of our interactome data. Gene Ontology (GO) analysis showed distinct functional GO categories including ion channels, cellular signaling molecules, apoptosis, ER function and mitochondrial function. We finally mapped the identified proteins using the STRING interaction database that further revealed functional relationships between the STIM1 complex proteins and significant changes during hypertrophic remodeling. These findings identify multiple specific functional complexes for STIM1 that are recruited during cardiac hypertrophy. These data support a model where STIM1 is a dynamic signal transducer that can interact with a diversity of proteins depending on the pathophysiological conditions.
Article Information
vol. 130 no. Suppl 2 A17732
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Ahyoung Lee;
- Changwon Kho;
- Ludovic Bénard;
- Antoine Chaanine;
- Roger Hajjar;
- Jean-Sébastien Hulot
- Cardiovascular Rsch Cntr, Icahn Sch of Medicine at Mount Sinai, New York, NY
Abstract 17784: The Genesips Project: an NHLBI-Sponsored induced Pluripotent Stem Cell (iPSC) Resource for the Study of Cardiovascular Diseases
Ivan Carcamo-Orive, Paige Cundiff, Hope Lancero, Mohammad Shahbazi, Fahim Abbasi, Sheena Abraham, Gerald Reaven, Sean Whalen, Sunita D’Souza, Gaurav Pandey, Achchhe Patel, Eric Schadt, Ihor Lemischka, Joshua Knowles, Thomas Quertermous
Circulation. 2014;130:A17784
Abstract
The study of complex cardiovascular disease (CVD) has been hampered by the lack of appropriate human cellular model systems. In response, the NHBLI sponsored the NextGen Consortium, which encompasses 9 independent efforts spanning the portfolio of NHLBI related phenotypes. The goals of the consortium include: 1. To develop and improve methods for large-scale production and characterization of induced pluripotent stem cell (iPSC) models for CVD; 2. To create a resource of iPSC lines from a large number of phenotypically and genotypically characterized individuals.
Our GENESiPS project is focused on insulin resistance (IR), a condition that affects 25-33% of the US population with serious health consequences including risk of type II diabetes and CVD. Although much is known about the physiological changes occurring during IR, little is known about the molecular pathways that drive the appearance of IR. Certain mature cell types as adipocytes, endothelial cells and skeletal muscle cells have been associated with the origin, maintenance and progression of IR. IPSCs offer an unprecedented opportunity of modeling human disease in vitro. We have created iPSC lines on insulin resistant and insulin sensitive patient groups with prior GWAS genotyping. Differentiation of these iPSCs to relevant cell types is providing the opportunity to correlate insulin sensitivity and high-density genetic variation data with specific cell-based profiling. We will validate our in vitro model and study the molecular pathways that define IR and its relationship to endothelial dysfunction.
Relevant to the larger scientific community the establishment of iPSC lines on over 150 individuals (3 to 6 clones per patient) that reflect the range of insulin resistance in the general population. The iPSC lines were created from erythroblasts using the non-integrative Sendai virus system, passaged to allow clearance of Sendai virus and growth in feeder free conditions. The lines have been extensively characterized for markers of pluripotency (Tra1-60), sample identity and genomic integrity. Through the NextGen consortium, these lines, as well as phenotypic and genome-wide genotyping data will be available to qualified investigators.
Article Information
vol. 130 no. Suppl 2 A17784
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Ivan Carcamo-Orive1;
- Paige Cundiff2;
- Hope Lancero1;
- Mohammad Shahbazi1;
- Fahim Abbasi1;
- Sheena Abraham1;
- Gerald Reaven1;
- Sean Whalen3;
- Sunita D’Souza4;
- Gaurav Pandey5;
- Achchhe Patel2;
- Eric Schadt6;
- Ihor Lemischka2;
- Joshua Knowles1;
- Thomas Quertermous1
- 1Stanford Sch of Medicine and Cardiovascular Institute, Stanford Univ, Stanford, CA
- 2Dept of Developmental and Regenerative Biology, Icahn Sch of Medicine at Mount Sinai, New York, NY
- 3Gladstone Institutes, Univ of California, San Francisco, CA
- 4D, Icahn Sch of Medicine at Mount Sinai, New York, NY
- 5Dept of Genetics and Genomic Sciences, Icahn Sch of Medicine at Mount Sinai, New York, NY
- 6Dept of Genomics and Multiscale Biology, Icahn Sch of Medicine at Mount Sinai, New York, NY
Abstract 17440: A Novel Aptamer-based Proteomic Technology for Cardiac Biomarker Discovery
Debby Ngo, Michelle J Keyes, Robert E Gerszten
Circulation. 2014;130:A17440
Abstract
Background: Current proteomic technologies have been slow to identify and validate new cardiac biomarkers. Chemically modified DNA-aptamers can serve as highly multiplexed immuno-like assays that offer the advantage of high-throughput for biomarker discovery.
Methods: We applied a novel aptamer-based technique (SOMAscan™) that measures > 1100 proteins to samples from individuals undergoing a “planned” myocardial infarction (PMI: alcohol ablation for hypertrophic cardiomyopathy), in which each individual serves as their own control. Serial blood samples were obtained before, 10 and 60 minutes after PMI. Patients undergoing elective diagnostic coronary angiography served as negative controls. Coronary sinus (CS) blood samples were collected concurrent with peripheral samples to determine potential myocardial origin of protein changes.
Results: There were 301 proteins that significantly changed in the peripheral blood post ablation in a PMI derivation cohort (n=15; p < 5.71E-5 , one-way ANOVA repeated measures), excluding proteins identified in the negative controls. Forty-three of these proteins were subsequently validated in a smaller PMI cohort (n=6; p < 1.66E-4, one-way ANOVA repeated measures), including known cardiac markers of injury such as troponin I (p= 5.24E-13) and CK-MB (p= 1.39E-10). Many protein changes detected are novel in the context of myocardial injury, including decreases in GDF-11 (p= 6.28E-5), a secreted factor shown to reverse age-related cardiac hypertrophy in mice and increases in PPIB (p= 2.30E-7), a cyclosporine-binding protein. Additionally, CS blood analysis confirmed 25 proteins as significantly changed after PMI, including 20 of the validated peripheral blood proteins (n=6; p < 5.71E-5, one-way ANOVA repeated measures). Furthermore, CS vs. peripheral blood comparison revealed 12 of the 301 proteins as significantly different (n=6; p < 5.71E-5, two-way ANOVA repeated measures), seven of which were enriched in the CS suggesting possible myocardial origin.
Conclusions: In pilot studies, an aptamer-based proteomic scan detected new biomarkers of myocardial injury. These findings motivate additional clinical studies in large, heterogeneous patient cohorts with spontaneous coronary syndromes.
Article Information
vol. 130 no. Suppl 2 A17440
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Pulmonary and Critical Care Div, Massachusetts General Hosp, Boston, MA
- 2Institute for Heart, Vascular and Stroke Care, Massachusetts General Hosp, Boston, MA
- 3Cardiovascular Div, Massachusetts General Hosp, Boston, MA
Abstract 15716: Non-Targeted Metabolomic Profiling Identifies Novel Markers of Insulin Resistance
John F O’Sullivan, Daniel J Cuthbertson, Robert E Gerszten
Circulation. 2014;130:A15716
Abstract
Background: Targeted metabolomic profiling has identified metabolite classes associated with insulin resistance (IR) and incident metabolic disease, including branched-chain (BCAAs) and aromatic amino acids (AAAs). Emerging non-targeted profiling methods, which facilitate screening of many-fold more metabolites, hold great promise for the identification of additional disease biomarkers and pathways. We developed a non-targeted workflow to determine differentially regulated metabolites in two groups that differed by the homeostatic model assessment of IR (HOMA-IR).
Methods: We analyzed fasting plasma from subjects with IR (HOMA-IR ≥ 2.6) (mean age = 67 ± 1.49; BMI = 28 ± 0.63; HOMA-IR = 7.94 ± 1.67) and normal subjects (HOMA-IR ≤ 1.85) (mean age = 68 ± 2.42; BMI = 26 ± 0.86; HOMA-IR = 1.13 ± 0.08), using HILIC chromatography coupled to an Agilent™ 6550 iFunnel Q-TOF mass spectrometer operated in positive ion mode. Batch recursive metabolite feature extraction was performed using MH Profinder software followed by differential metabolite analysis and identification using MPP software.
Results: We identified 56 differentially abundant compounds [p < 0.05; unpaired t-test with Benjamini-Hochberg FDR (B-H FDR) correction] in a derivation cohort (n=15 IR; n=15 control) and validated 28 of these compounds in a similarly sized independent cohort (p <0.05, BH-FDR corrected). As expected, we confirmed compounds known to be associated with IR, including the BCAA valine (p=2.9E-3), the AAA tryptophan (p=1.01E-2), and a hexanoylcarnitine (p=4.8E-2). In addition, we made putative identifications of novel compounds that were significantly elevated in IR subjects in both cohorts, including aminooctanoic acid (+41%, p=3.68E-3), methylhistidine (+92%, p=4.78E-3), and methylinosine (+51%, p=3.29E-2). Novel compounds that were significantly decreased in IR subjects included isovalerylglycine (-48%, p=2.03E-2).
Conclusions: We have developed a non-targeted metabolite profiling platform and detected novel compounds associated with IR. Future work includes unambiguous identification and quantification of these metabolites using isotope-labeled standards, as well as evaluation of these markers in large, heterogeneous clinical populations.
Article Information
vol. 130 no. Suppl 2 A15716
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Cardiovascular Rsch Cntr, Massachusetts General Hosp, Boston, MA
- 2Molecular Plant Sciences, Washington State Univ, Pullman, WA
Abstract 13301: Methylome-wide Association With Soluble Cell Adhesion Molecules Among Monozygotic Twins
Yan Sun, Jack Goldberg, Dean P Jones, Viola Vaccarino
Circulation. 2014;130:A13301
Abstract
Introduction: Inflammation plays a critical role in the pathogenesis of cardiovascular disease. Epigenetic mechanisms, including DNA methylation (DNAm), is critical in the regulation of inflammatory genes, and can be influenced by inflammation. The soluble form of cell adhesion molecules, including vascular adhesion molecule 1 (sVCAM1), intercellular adhesion molecule 1 (sICAM1), and P-selectin (sP-selectin), are established biomarkers for inflammation and endothelial function, and are linked to cardiovascular events.
Methods: To identify epigenetic markers associated with inflammation and endothelial function, we conducted a methylome-wide association study and investigated over 480,000 DNAm sites of peripheral blood cells from 140 monozygotic (MZ) middle-aged male twins from the Emory Twin Study.
Results: Using two randomly selected subsets consisting of unrelated subjects, we identified and replicated 69 and 23 DNAm sites significantly associated with sVCAM1, and sICAM1 respectively, adjusted for multiple testing, but none for sP-selectin. All 23 sICAM1-associated DNAm sites were also associated with sVCAM1, including sites on gene ANKRD11 (P=1.51х10-21, 2.62х10-20), KDM2B (P=1.52х10-21, 9.13х10-17), CAPS (P=2.81х10-20, 3.17х10-18), and CUX1 (P=7.63х10-20, 2.84х10-19). They jointly explained 54% and 40% of variance in sVCAM1 and sICAM1 respectively. Two DNAm sites, located on UNC5D and TMEM125, were also significant comparing MZ twins who were phenotypically discordant for both sICAM1 (P=1.79х10-7, 2.78х10-6) and sVCAM1 (P=1.70х10-9, 1.71х10-7).
Conclusions: These results suggest that sVCAM1 and sICAM1, but not sP-selectin, may share common pathophysiology in inflammation and endothelial function via an epigenetic mechanism in leukocytes. In addition, the epigenetic association with inflammation may be driven by unshared environmental exposures.
Article Information
vol. 130 no. Suppl 2 A13301
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Epidemiology, Emory Univ, Atlanta, GA
- 2Epidemiology, Univ of Washington, Seattle, WA
- 3Medicine, Emory Univ, Atlanta, GA
Abstract 11249: Differential Cardiac MicroRNA Expression Predicts the Clinical Course in Human Enterovirus Cardiomyopathy
Wolfgang Poller
Circulation. 2014;130:A11249
Abstract
Background: Regarding disease pathogenesis, it has become evident that confinement to analysis of protein-coding regions of the genome is incomplete since many noncoding variants are associated with important human diseases
Objectives: Identification of novel molecular predictive markers for the course of human enterovirus (CVB3) cardiomyopathy, and of noncoding elements in the genome influencing the grossly different antiviral capacity of individual patients.
Methods: Transcriptome mapping of CVB3 cardiomyopathy patients revealed distinctive cardiac microRNA (miR) patterns associated with either spontaneous virus clearance and recovery (CVB3-ELIM), or virus persistence and progressive clinical deterioration (CVB3-PERS). Profiling of protein-coding genes and 754 miRs in endomyocardial biopsies (EMBs) of test cohorts was performed at their initial presentation, and those spontaneously eliminating the virus were compared to those with virus persistence on follow-up.
Results: Initial miR profiling revealed highly significant differences in the cardiac levels of 16 miRs, but not in protein-coding genes. Eight miRs were strongly induced in CVB3-PERS only (miRs 135b, 155, 190a, 422a, 489, 590, 601, 1290), but undetectable in CVB3-ELIM or controls. Remarkably, four of the miRs deregulated in CVB3-PERS vs. CVB3-ELIM are primate-specific, and thus do not exist in murine models of human CVB3 cardiomyopathy. Further evaluation of this primary distinctive miR pattern in validation cohorts, and multivariate statistical analysis, led to the definition of a secondary simplified predictive miR profile suggested for future clinical use.
Conclusions: A clinical application of these data is cardiac miR profiling to assess the risk of virus persistence and progressive clinical deterioration in CVB3 cardiomyopathy. Patients at risk are eligible for immediate antiviral therapy to minimize irreversible cardiac damage. A basic science aspect is the failure of protein-coding gene profiling to identify significant predictors, suggesting that individual antiviral capacity is strongly influenced and reflected by noncoding miR transcripts some of which may constitute new therapeutic targets.
Article Information
vol. 130 no. Suppl 2 A11249
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Wolfgang Poller
- Cardiology and Pneumology CBF, Charite – Universitätsmedizin Berlin, Berlin, Germany
Abstract 17697: Direct Physician Reporting Reduces Radiation Exposure in Pediatric Cardiac Catheterizations
George T Nicholson, Dennis W Kim, Robert N Vincent, Kevin Gao, Virginia Balfour, Christopher J Petit
Circulation. 2014;130:A17697
Abstract
Objectives: To quantify radiation dose in children undergoing cardiac catheterization and determine the impact of increased reporting transparency on total radiation exposure.
Background: Cardiac catheterization (cath) can result in significant radiation exposure in children. There is growing interest in quantifying and reducing radiation exposure in pediatric cath procedures. Our center joined a national registry in 2011 which required reporting of radiation exposure. In 2013, our center began participating in a second registry in which physicians directly entered radiation data. No specific educational or interventional radiation measures were taken during the study periods.
Methods/Results: We reviewed up to 20 cases across 3 time periods in each of 4 categories: post-heart transplant annual cath, unilateral pulmonary artery (PA) stent placement, pre-Fontan cath, and pre-Glenn cath. The 3 Eras were defined as: Era 1, 1/2009 – 1/2011; Era 2, 1/2011 – 9/2013; and Era 3, 9/2013 – 5/2014. In Era 3, the physician performing the cath was responsible for reporting the radiation data. Patients in each case category were matched for age, weight, and body surface area across 3 time periods.
Across the 3 Eras, there were significant decreases in cumulative air KERMA (mGy) among the unilateral PA stent placement (p < 0.01) and pre-Glenn evaluation (p = 0.01) cases. Although the decrease among the annual evaluation (p = 0.21) and pre-Fontan (p = 0.43) cases were not significant, the overall trend was a decrease in radiation exposure. From Era 2 to 3, there were significant decreases in dose area product (cGy·m2) in the annual evaluation (p = 0.01), PA stent placement (p = 0.02), and pre-Glenn (p = 0.01) cases. Dose area product information was not available in Era 1. In Era 1, 0 cases (0%) had a frame rate change during any individual cath, while in Era 2, 9 cases (11.25%), and in Era 3, 15 cases (23.4%) had frame rate changes (p < 0.01).
Conclusions: Increased physician awareness of radiation exposure significantly reduces radiation dose across a variety of procedures. This is seen not only by the overall reduction in radiation across case types, but also as the frame rate was more frequently changed during individual cases, indicating a change in physician behavior and practice.
Article Information
vol. 130 no. Suppl 2 A17697
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- George T Nicholson;
- Dennis W Kim;
- Robert N Vincent;
- Kevin Gao;
- Virginia Balfour;
- Christopher J Petit
- Cardiology, Children’s Healthcare of Atlanta/Emory Univ, Atlanta, GA
Abstract 12450: Myocardial Fibrosis Burden Predicts Left Ventricular Ejection Fraction and is Modified by Age and Steroid Treatment Duration in Duchenne Muscular Dystrophy
Animesh Tandon, Chet R Villa, Kan N Hor, John L Jefferies, Zhiqian Gao, Jeffrey A Towbin, Brenda L Wong, Wojciech Mazur, Robert J Fleck, Joshua J Sticka, Dudley W Benson, Michael D Taylor
Circulation. 2014;130:A12450
Abstract
Background: Patients with Duchenne muscular dystrophy (DMD) typically exhibit progressive cardiac and skeletal muscle dysfunction, and are commonly treated with corticosteroids to prolong ambulation. Myocardial fibrosis and steroid treatment may modulate the progression of cardiac dysfunction in DMD patients. We aimed to longitudinally characterize the impact of myocardial fibrosis and steroid treatment on the progression of cardiac dysfunction using cardiac magnetic resonance (CMR) in a large DMD cohort.
Methods: Serial CMR studies performed on DMD patients were reviewed for LVEF and late gadolinium enhancement (LGE) status, a marker for myocardial fibrosis. LVEF was modeled by examining effects of patient age, steroid treatment duration, LGE status, and myocardial fibrosis burden, as assessed by the number of LGE positive (LGE+) LV segments.
Results: We analyzed 469 CMR studies from 99 DMD patients with ≥4 CMRs. Patient age at time of CMR ranged from 6.6 to 29.4 (median 12.3) years. There were 146 (31.1%) LGE+ studies and 59 studies (12.6%) that demonstrated depressed LVEF (LVEF<55%). An age-only model demonstrated that LVEF declined 0.58±0.10%/yr (p<0.0001, r2=0.067). Univariate modeling showed significant associations between LVEF and steroid treatment duration, presence of LGE, and number of LGE+ LV segments; multivariate modeling showed that LVEF declined by 0.93±0.09% for each LGE+ LV segment (p<0.0001, r2=0.171), while age and steroid treatment duration were no longer significant. The number of LGE+ LV segments increased with age by 1.2 segments/year (95% confidence interval 1.1-1.2), and steroid treatment partially mitigated this increase (interaction term β=-0.01±0.005, p=0.010).
Conclusions: Progressive myocardial fibrosis, as imaged by LGE on CMR, is a strong marker for the decline in LVEF in DMD patients. Steroid treatment partially attenuates the age-related increase in myocardial fibrosis burden.
Article Information
vol. 130 no. Suppl 2 A12450
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Animesh Tandon1;
- Chet R Villa1;
- Kan N Hor2;
- John L Jefferies1;
- Zhiqian Gao1;
- Jeffrey A Towbin1;
- Brenda L Wong3;
- Wojciech Mazur4;
- Robert J Fleck5;
- Joshua J Sticka1;
- Dudley W Benson6;
- Michael D Taylor1
- 1The Heart Institute, Cincinnati Children’s Hosp Med Cntr, Cincinnati, OH
- 2The Heart Cntr, Nationwide Children’s Hosp, Columbus, OH
- 3Div of Neurology, Cincinnati Children’s Hosp Med Cntr, Cincinnati, OH
- 4The Heart and Vascular Cntr, The Christ Hosp, Cincinnati, OH
- 5Dept of Radiology, Cincinnati Children’s Hosp Med Cntr, Cincinnati, OH
- 6Herma Heart Cntr, Children’s Hosp of Wisconsin, Milwaukee, WI
Abstract 12075: Whole-Genome Sequencing at 10-Days of Life in Perinatal Long-QT Syndrome Yields New Insights Into Disease Pathogenesis
James R Priest, Charles Gawad, Kris Kahlig, Scott Ceresnak, Lindsey Malloy-Walton, Kyla Dunn, Megan P Grove, Marco Perez, Katsuhide Maeda, Anne Dubin, Luiz Belardinelli, Rich Chen, Tom Quertermous, Stephen Quake, Euan A Ashley
Circulation. 2014;130:A12075
Abstract
Introduction: Perinatal LQTS represents a severe form of long-QT syndrome with poor outcomes and early genotype-specific treatment is limited by the 2 month turnaround time of standard panel genetic testing.
Hypothesis: We aimed to provide a molecular diagnosis within a clinically actionable timeframe.
Methods: We performed rapid CLIA-certified whole genome sequencing on two infants with perinatal long-QT syndrome delivering a molecular diagnosis at 10-days of life. Whole cell patch clamping and single cell genotyping were also performed.
Results: In Case #1 we discovered a previously characterized variant in KCNH2 which was paternally inherited, however whole genome sequencing provided an unbiased assessment of the entire catalog of human genes revealing a second maternally inherited modifier variant in RNF207.
In Case #2 we discovered a novel mutation leucine replacing valine (V1762L) at residue 1762 in the SNC5A sodium channel. Whole-cell patch clamping experiments show the V1762L mutation causes a profound defect in late sodium current ~4.5 fold greater than the wild-type channel. A single cell analysis demonstrated that the mutation was present in the genome of only 3 of 36 individually isolated and genotyped patient cells, suggesting mosaicism. Conspicuously, standard panel genetic testing was negative.
Conclusions: We report here the earliest molecular diagnoses of LQTS, and demonstrate that rapid whole genome sequencing may be fruitfully applied to perinatal LQTS. In case #1 we hypothesize that a polygenic inheritance may explain the early and severe perinatal presentation, and have identified a putative modifier gene for LQTS in RNF207. The observation of mosaicism in case #2 suggests that in studies of inherited disease, mosaicism represents a common mechanism by which causal variation may be missed.
Article Information
vol. 130 no. Suppl 2 A12075
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- James R Priest1;
- Charles Gawad1;
- Kris Kahlig2;
- Scott Ceresnak1;
- Lindsey Malloy-Walton1;
- Kyla Dunn3;
- Megan P Grove4;
- Marco Perez4;
- Katsuhide Maeda5;
- Anne Dubin1;
- Luiz Belardinelli2;
- Rich Chen6;
- Tom Quertermous4;
- Stephen Quake7;
- Euan A Ashley4
- 1Dept of Pediatrics (Cardiology), Stanford Univ Med Cntr, Stanford, CA
- 2Cardiovascular Therapeutics, Gilead Inc, Fremont, CA
- 3Lucile Packard Children’s Hosp Heart Cntr, Stanford Univ Med Cntr, Stanford, CA
- 4Div of Cardiovascular Medicine, Stanford Univ Med Cntr, Stanford, CA
- 5Div of Cardiothoracic Surgery, Stanford Univ Med Cntr, Stanford, CA
- 6none, Personalis Corp, Menlo Park, CA
- 7Dept of Bioengineering, Stanford Univ Sch of Medicine, Stanford, CA
Abstract 17568: Arrhythmogenic Myocardial Remodeling in Adults with Surgically Corrected Tetralogy of Fallot
Heiko Schneider, Oliver Monfredi, Lucy Murfitt, Hayley Bennett, Ian P Temple, David Knight, Ronan O’Cualain, Halina Dobrzynski, Julian Selley, Parveen Sharma, George Hart, Ashraf Kitmitto, Andreas J Hoschtitzky, Mark R Boyett, Vaikom S Mahadevan
Circulation. 2014;130:A17568
Abstract
Introduction: Adults after repair of Tetralogy of Fallot (ToF) have an increased risk of heart failure, arrhythmias and sudden death.
We hypothesize that electrophysiological remodeling of the right atrium (RA), right ventricle (RV) and right ventricular outflow tract (RVOT) is responsible.
Methods: We collected RA, RV and RVOT biopsies from 10 ToF patients (6 male) at the time of pulmonary valve replacement. Biopsies were also taken from 9 control patients (4 male) with left ventricular outflow tract obstruction without evidence of septal defects, previous arrhythmia or right-sided heart disease. We performed: (i) extracellular matrix (ECM) quantification; (ii) RT-qPCR to quantify mRNA for ion channels, transporters, connexins, inflammatory markers and ECM constituents; (iii) proteomics (liquid chromatography and mass spectrometry (MS)) on 6 patients per group to quantify ~1600 proteins; and (iv) Western Blot for selected proteins.
Results: Mean age at surgery was 32±4 and 22±2 years in the ToF and control groups respectively. There were significant changes in the relative abundance of mRNAs for ion channels, adrenergic receptors, ECM, and heart failure markers (ANP and BNP) in ToF patients compared with controls. MS highlighted over 300 significant differences, specifically changes in proteoglycans and downregulation of mitochondrial respiratory complexes 1 and 3 in ToF patients compared with controls. No significant difference in fibrosis on histological analysis between the two groups was seen. A selection of changes is displayed in Table 1.
Conclusion: The RA and RVOT undergo significant remodeling in ToF. Changes in ion channels, transporters and Ca2+-handling (especially SERCA2A, RYR2, Calsequestrin) as well as gap junction proteins (Cx43) are likely to alter ionic currents, with increased arrhythmia risk. This remodeling is similar to changes in heart failure and ischemia-reperfusion and may contribute to the arrhythmias seen in ToF.
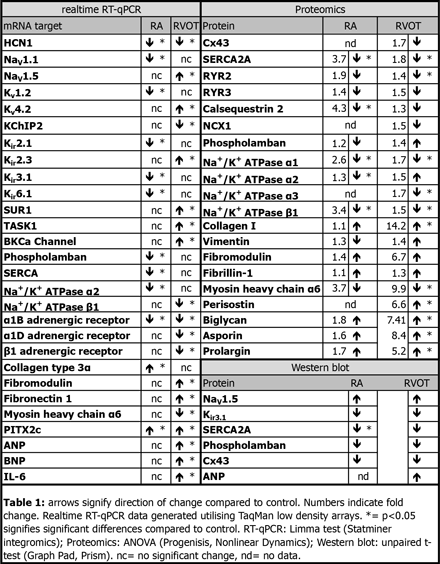
Article Information
vol. 130 no. Suppl 2 A17568
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Heiko Schneider1;
- Oliver Monfredi1;
- Lucy Murfitt1;
- Hayley Bennett1;
- Ian P Temple1;
- David Knight2;
- Ronan O’Cualain2;
- Halina Dobrzynski1;
- Julian Selley2;
- Parveen Sharma3;
- George Hart1;
- Ashraf Kitmitto1;
- Andreas J Hoschtitzky4;
- Mark R Boyett1;
- Vaikom S Mahadevan5
- 1Institute of Cardiovascular Sciences, Univ of Manchester, Manchester, United Kingdom
- 2Faculty of Life Sciences, Univ of Manchester, Manchester, United Kingdom
- 3Institute of Translational Medicine, Univ of Liverpool, Liverpool, United Kingdom
- 4Manchester Heart Cntr, Central Manchester Univ Hosps NHS Foundation Trust, Manchester, United Kingdom
- 5Manchester Heart Cntr, Central Manchester Univ Hosp NHS Foundation Trust, Manchester, United Kingdom
Abstract 16607: Pediatric-Onset Disease Does Not Herald Adverse Clinical Course in Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy
Anneline S te Riele, Cynthia A James, Abhishek C Sawant, Brittney Murray, Crystal Tichnell, Ryan Tedford, Jane Crosson, Daniel P Judge, Hugh Calkins, Harikrishna Tandri
Circulation. 2014;130:A16607
Abstract
Background: In most genetic cardiomyopathies, early disease onset heralds adverse clinical outcome. However, the clinical attributes and disease course in pediatric cases with Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy (ARVD/C) are largely unknown.
Objective: To delineate and compare the clinical characteristics, genetics, and outcome of pediatric-onset ARVD/C.
Methods: We obtained detailed phenotypic, genetic, and outcome data of 316 subjects who presented alive and fulfilled diagnostic Task Force Criteria (TFC) for ARVD/C at last follow-up. Patients were grouped into pediatric (diagnosis at <18 years) and adult (diagnosis at ≥ 18 years) ARVD/C. Clinical outcomes regarding sustained ventricular arrhythmia, cardiac transplantation, and death were ascertained.
Results: Among 316 definite ARVD/C cases, 43 (14%) were diagnosed prior to the age of 18 years. Pediatric cases were 22 (51%) males, with a mean age of 15.4 ± 1.8 years at time of diagnosis. Compared to adult cases, pediatric cases were disproportionately mutation carriers (77% vs 59%, p=0.029), but not more likely to carry multiple mutations (3% vs 4%, p=0.729), or to be probands (72% vs 72%, p=0.968). There were no other differences in demographic characteristics or in any domain of the TFC. During 6.2 (IQR 2.4-11.3) years follow-up, 26 (61%) pediatric cases experienced a sustained ventricular arrhythmia, 1 (2%) had cardiac transplantation, and 2 (5%) died (1 heart failure and 1 sudden cardiac death). There were no differences in survival free from sustained ventricular arrhythmia (p=0.460), cardiac transplantation (p=0.887) or death (p=0.196) between pediatric and adult cases.
Conclusion: Pediatric ARVD/C patients are disproportionately mutation carriers. All other clinical characteristics are similar between pediatric and adult cases. Over more than 6 years follow-up, arrhythmic, heart failure, and mortality outcomes are the same in pediatric and adult ARVD/C patients.
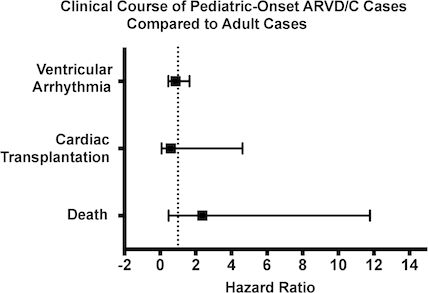
Article Information
vol. 130 no. Suppl 2 A16607
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Anneline S te Riele;
- Cynthia A James;
- Abhishek C Sawant;
- Brittney Murray;
- Crystal Tichnell;
- Ryan Tedford;
- Jane Crosson;
- Daniel P Judge;
- Hugh Calkins;
- Harikrishna Tandri
- Cardiology, Johns Hopkins Univ, Baltimore, MD
Abstract 13312: Risk Factors for the Development of Tachyarrhythmia Following the Norwood Procedure in the Single Ventricle Reconstruction Trial
Abstract
Background: Tachyarrhythmias are an important contributor to morbidity following the Norwood procedure for single ventricle congenital heart disease. The purpose of this study was to determine what demographic, preoperative, or operative factors may be associated with the development of postoperative tachyarrhythmias in this population.
Methods: We performed a retrospective analysis of data collected prospectively through the multicenter Pediatric Heart Network Single Ventricle Reconstruction Trial for infants undergoing a Norwood procedure in 2005-2008. Our primary outcome of interest was any documented tachyarrhythmia requiring treatment or intervention during the inpatient postoperative stay. Subjects with a documented preoperative tachyarrhythmia (n=15) were excluded. After performing univariate chi-square analyses on a variety of candidate factors, we conducted multivariate stepwise logistic regression, including spline terms for nonlinearly associated variables, to calculate the adjusted odds ratios for the final variables.
Results: Of 529 subjects, 108 (20%) had at least one documented tachyarrhythmia, with 11 having more than one. Tachyarrhythmias included: 77 supraventricular tachycardia, 23 junctional ectopic tachycardia, 10 atrial flutter, 7 ventricular tachycardia, and 2 atrial fibrillation. In the final multivariate model (c-statistic=0.65), significant factors associated with arrhythmia included receiving a modified Blalock-Taussig shunt, use of ultrafiltration, and age. (Table)
Conclusions: Tachyarrhythmias are common following the Norwood procedure and are associated with the modifiable risk factors of shunt type and use of ultrafiltration. In addition, when surgery is performed between 8 and 20 days of age, older age is associated with a decreasing risk for tachyarrhythmia.

Article Information
vol. 130 no. Suppl 2 A13312
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Matthew Oster1;
- Shan Chen2;
- David Pober2;
- Yaniv Bar-Cohen3;
- Matthew Brothers1;
- Nicole Cain4;
- Steve Colan5;
- Richard Czosek6;
- Jamie Decker7;
- David Gamboa8;
- Salim Idriss9;
- Joel Kirsh10;
- Martin LaPage11;
- Richard Ohye12;
- Elizabeth Radojewski13;
- Maully Shah14;
- Eric Silver15;
- Anoop Singh16;
- Joel Temple17;
- John Triedman5;
- Jonathan Kaltman18
- 1Sibley Heart Cntr Cardiology, Children’s Healthcare of Atlanta, Atlanta, GA
- 2Statistics, New England Rsch Institutes, Inc., Watertown, MA
- 3Cardiology, Children’s Hosp Los Angeles, Los Angeles, CA
- 4Cardiology, MUSC Children’s Hosp, Charleston, SC
- 5Cardiology, Boston Children’s Hosp, Boston, MA
- 6The Heart Institute, Cincinnati Children’s Hosp Med Cntr, Cincinnati, OH
- 7Cardiology, Johns Hopkins All Children’s Hosp, Saint Petersburg, FL
- 8Pediatric Cardiology, Univ of Utah / Primary Children’s Hosp, Salt Lake City,, UT
- 9Pediatric Cardiology, Duke Univ, Durham, NC
- 10Dept of Paediatrics, Hosp for Sick Children & Univ of Toronto, Toronto, Canada
- 11Pediatrics and Communicable Diseases, Div of Cardiology, Univ of Michigan, Ann Arbor, MI
- 12Cardiac Surgery, Univ of Michigan C. S. Mott Children’s Hosp, Ann Arbor, MI
- 13Cardiology, Hosp for Sick Children, Toronto, Canada
- 14Cardiology, The Children’s Hosp of Philadelphia, Philadelphia, PA
- 15Pediatrics, New York-Presbyterian/Morgan Stanley Children’s Hosp, Columbia Univ Med Cntr, New York, NY
- 16Pediatric Cardiology, Med College of Wisconsin, Milwaukee, WI
- 17Cardiology, Nemours Cardiac Cntr, Wilmington, DE
- 18Div of Cardiovascular Sciences, The National Heart, Lung, and Blood Institute, Bethesda, MD
Abstract 13544: Differences in the Population of Muscular Dystrophy Patients With Respect to Ventricular Tachycardia, Heart Failure and Use of Implantable Cardioverters Defibrillators
Ann R Punnoose, Jonathan Kaltman, Christopher Spurney
Circulation. 2014;130:A13544
Abstract
Introduction: As the treatment of the respiratory and skeletal muscle complications associated with muscular dystrophy (MD) has improved, males with MD are living longer and are more likely to develop cardiac disease. The impact of sudden cardiac death related to ventricular arrhythmias and heart failure and the indications for ICD implantation are not well established.
Hypothesis: We evaluated the hypothesis that VT is a significant contributor to mortality in hospitalized patients with MD and this is improved with ICD implantation.
Methods: We searched the Pediatric Health Information System (PHIS) database for males with discharge diagnosis codes of MD and different combinations of heart failure (HF), ventricular arrhythmias (VT) and ICD placement between 2003 and 2013.
Results: There were significant differences in the mean length of stay and mean age between MD patients with HF and/or VT versus all hospitalized male MD patients (p<0.05). Out of a total of 76 deaths in MD patients, 32% (n=24) were related to HF. Of those, 30% (n=7) also had a diagnosis of VT. Isolated VT without HF was only 1% (n=42) of admissions and the cause of death in 2 patients. However, ICD’s were implanted in 12% (n=5) of MD/VT patients compared to 5% (n=5) of MD/HF/VT patients. No deaths were seen in patients with ICD’s. Only 1 ICD was placed prior to 2009.
Conclusions: VT was a frequent cause of death in hospitalized male patients with MD and HF, however isolated VT without HF was not. No deaths were found in the small number of patients with ICD’s. Further prospective studies are needed to validate the importance of ICD use in MD patients
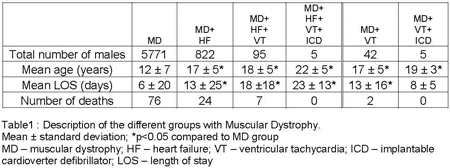
Article Information
vol. 130 no. Suppl 2 A13544
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Pediatric Cardiology, Children’s National Med Cntr, Washington, DC
- 2Pediatric Cardiology, The National Institute of Health and the Children’s National Med Cntr, Washington, DC
Abstract 20594: Variation of Radiation Usage and Current Practice in the Pediatric Cardiac Catheterization Laboratory – A Multicenter Study by the CCISC (Congenital Cardiovascular Interventional Study Consortium)
Daisuke Kobayashi, Thomas J Forbes
Circulation. 2014;130:A20594
Abstract
Introduction: Reduction of radiation dosage is an important task of quality improvement in pediatric cardiac catheterization laboratories. Comparison of data with a benchmark allows intra and inter-institutional comparisons to be made. To date, institutional variation of catheterization practices focusing on radiation dosage has not been well described.
Hypothesis: We hypothesized that radiation dosage significantly vary between institutions and there may be significant effect of frame rate on radiation dosage.
Methods: This was a multicenter observational study of children undergoing catheterizations in pediatric laboratories. Utilizing normalized air Kerma area product (PKA) by body weight (PKA/BW, μGym2/kg) as a standardized measure, institutional variation and trend of radiation dosage from 2009 to 2013 were analyzed. Cases were broken down into interventional, diagnostic, and transplant right ventricular transplant biopsy. These cases were further categorized into high (30 frame/sec for cine and 15 for fluoroscopy) and low (15 and 7.5, respectively) frame rate groups and the effect of frame rate on radiation dosage was assessed.
Results: Among 8267 procedures from 16 institutions, PKA/BW was significantly variable between institutions (p<0.001). There was a gradual decrease in the annual median PKA/BW of diagnostic and interventional procedures over 5 years, despite no significant change in fluoroscopic time. High frame rate group had significantly higher PKA/BW (p<0.001) in diagnostic and interventional catheterizations.
Conclusions: PKA/BW appeared to be a useful standardized measure of radiation dose to compare between institutions. Significant variation of radiation dosage suggests a need of quality control in this field. Use of lower frame rates significantly lowered radiation dosages for similar procedural types. A concerted effort should be made to lower the frame rates in labs with consistently higher radiation usage.
Article Information
vol. 130 no. Suppl 2 A20594
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Daisuke Kobayashi;
- Thomas J Forbes
- Div of Cardiology, Carman and Ann Adams Dept of Pediatrics, Children’s Hosp of Michigan, Wayne State Univ Sch of Medicine, Detroit, MI
Abstract 19807: Pre-Superior Cavopulmonary Anastomosis Cardiac Catheterization at Altitude
Michael V Di Maria, Thomas E Fagan, Neil Wilson, Max B Mitchell, David N Campbell, James Jaggers, Eduardo da Cruz, D. Dunbar Ivy, Adel K Younoszai
Circulation. 2014;130:A19807
Abstract
Introduction: Infants with single ventricle physiology typically undergo cardiac catheterization prior to superior cavopulmonary anastomosis (SCPA) to assess operative suitability. Predictors of poor outcome at sea level include elevated central venous pressure (CVP), transpulmonary gradient (TPG), pulmonary vascular resistance (PVR) and pulmonary artery (PA) size. Living at higher altitude has vasoconstrictive effects on the pulmonary vasculature, and a prior study suggested that higher PA pressure may predict worse outcomes. The goal of this study was to determine which elements of the pre-SCPA catheterization were useful in predicting successful Fontan operation at altitude.
Methods: A retrospective review revealed 150 patients who underwent pre-SCPA catheterization over a 10-year period. Pre-SCPA catheterization data were abstracted and subjects were grouped by progression to Fontan vs. aborted palliation, heart transplant or death. Statistical analysis included Wilcoxon Rank Sum tests, uni-variable logistic regression and receiver operator characteristic (ROC) curve analysis.
Results: Differences between groups at cardiac catheterization are summarized in Table 1. Logistic regression showed that larger PA diameter was protective; left PA: OR: 0.73, 95% CI: 0.6-0.9, p = 0.01; right PA: OR:0.73, 95% CI: 0.6-0.9, p = 0.02. ROC analysis defined thresholds for minimum left PA and right PA diameter of <4mm for poor outcome (area under the curve of 0.68 and 0.67, respectively).
Conclusions: Our data suggest that pulmonary arterial size, more so than measured pressure or resistance, influences ability to achieve Fontan palliation at higher altitude. We hypothesize that this may be a feature of differences in pulmonary arterial growth. An alternative approach to evaluating pulmonary arterial morphology during palliation, such as cross-sectional imaging, may help optimize individual patient hemodynamics and provide better predictors of outcome.
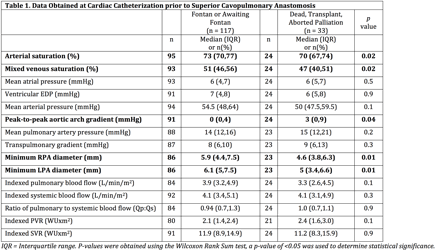
Article Information
vol. 130 no. Suppl 2 A19807
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Michael V Di Maria1;
- Thomas E Fagan1;
- Neil Wilson1;
- Max B Mitchell2;
- David N Campbell2;
- James Jaggers2;
- Eduardo da Cruz1;
- D. Dunbar Ivy1;
- Adel K Younoszai1
- 1Pediatrics, Children’s Hosp Colorado, Univ of Colorado Sch of Medicine, Aurora, CO
- 2Surgery, Children’s Hosp Colorado, Univ of Colorado Sch of Medicine, Aurora, CO
Abstract 19567: Severe Pulmonary Insufficiency is Common > 10 Years Following Balloon Pulmonary Valvuloplasty for Isolated Pulmonary Valve Stenosis
Rajiv Devanagondi, Dan Peck, Janaki Sagi, Janet Donohue, Sunkyung Yu, Sara K Pasquali, Aimee Armstrong
Circulation. 2014;130:A19567
Abstract
Introduction: While balloon pulmonary valvuloplasty (BPV) for isolated pulmonary stenosis successfully relieves obstruction acutely, long term outcomes remain unclear, with only one study to date exclusively reporting >10 year outcomes (n=48 subjects). We evaluated the long-term incidence of ≥moderate pulmonary insufficiency (PI) and reintervention.
Methods: Patients undergoing BPV at our institution from 1982-2002 for isolated pulmonary valve stenosis (n=211) were eligible for inclusion. Long-term follow-up data were available for 103 patients (primarily those followed at our center). Incidence of ≥moderate PI and reintervention were reported and risk factors for ≥moderate PI assessed in univariate and multivariable analysis.
Results: Of 103 included patients, age at initial BPV was 0.7 yrs (range 1d-42.2 years), BSA 0.38 m2 (range 0.14-1.99 m2), peak cath gradient 65 mmHg (range 31-169 mmHg) and 23% had critical pulmonary stenosis. In the first 10 years after BPV, 16% had surgical pulmonary valvotomy, transannular patch, or BT shunt and 2% died. Of the remaining patients with >10-year follow up data (median follow-up 15.1 years, range 10.1-26.3 years), 62 had a recent echocardiogram. Of these patients, 37% had moderate and 23% had severe PI; RV dilatation was mild in 32% and moderate in 10%, while 5% had mild RV systolic dysfunction. Three patients had pulmonary valve replacement 16.8-22.2 yrs following BPV due to severe PI. In univariate analysis, critical pulmonary stenosis, younger age, smaller BSA, and smaller pulmonary annulus at the time of BPV, as well as greater pre-BPV cath gradient were associated with ≥moderate PI (all p<0.05). There was no significant association between balloon:annulus ratio and ≥moderate PI. In multivariable analysis, BSA <0.3 m2 remained significantly associated with ≥moderate PI (OR 6.4, 95% CI 1.2-33.6; p=0.03).
Conclusions: In the largest study to date of > 10 year outcomes following BPV nearly ¼ of patients developed severe PI. Although few patients have required pulmonary valve replacement to date, close follow up is necessary. Patients with younger age, lower BSA, and/or more severe pulmonary stenosis at the time of BPV have a greater risk of significant PI and should be counseled regarding the long term risks.
Article Information
vol. 130 no. Suppl 2 A19567
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Rajiv Devanagondi;
- Dan Peck;
- Janaki Sagi;
- Janet Donohue;
- Sunkyung Yu;
- Sara K Pasquali;
- Aimee Armstrong
- Div of Pediatric Cardiology, Univ of Michigan, Ann Arbor, MI
Abstract 19583: Autonomous and Non-Autonomous Defects Underlie Hypertrophic Cardiomyopathy in BRAF-Mutant hiPSC-Derived Cardiomyocytes
Rebecca Josowitz, Sonia Mulero-Navarro, Christine Falce, Ninette Cohen, Erik M Ullian, Lauren A Weiss, Katherine A Rauen, Eric A Sobie, Bruce D Gelb
Circulation. 2014;130:A19583
Abstract
Hypertrophic cardiomyopathy (HCM) is a pathological disorder predominantly due to mutations in sarcomeric components. Germline mutations in BRAF cause a developmental syndrome called cardio-facio-cutaneous syndrome (CFCS), in which 40% of patients also develop HCM. Since the role of the RAS/MAPK pathway in HCM is still unclear, we generated a human induced pluripotent stem cell model for CFCS from three patients with activating BRAF T599R or Q257R mutations. In order to examine hiPSC-derived cell-type specific phenotypes and cellular interactions underpinning HCM, we generated a method to purify cardiomyocytes and non-cardiomyocytes simultaneously by cell sorting based on SIRPα and CD90 expression. Purified BRAF-mutant SIRPα+/CD90- cells were >95% cardiomyocytes, displayed cellular hypertrophy with pro-hypertrophic gene expression, and dysregulation of the RAS/MAPK pathway. BRAF-mutant cardiomyocytes also displayed intrinsic calcium handling defects, including increased calcium transient irregularity and increased stored calcium within the sarcoplasmic reticulum. In addition, purified BRAF-mutant SIRPα-/CD90+ cells, which were fibroblast-like, displayed activation of the RAS/MAPK pathway and exhibited a pro-fibrotic phenotype. Cross-culture studies revealed that BRAF-mutant fibroblast-like cells critically modulate cardiomyocyte hypertrophy through TGFβ paracrine signaling, as TGFβ inhibition prevented cardiomyocyte hypertrophy induced by BRAF-mutant fibroblast-like cells. Additionally, inhibition of RAS/MAPK signaling was capable of rescuing BRAF-mutant cardiomyocyte hypertrophy and cardiomyocyte-intrinsic calcium handling abnormalities. Thus, we show for the first time that cell autonomous and non-autonomous defects underlie RASopathy-associated HCM. As fibroblast activation has been documented previously in sarcomeric HCM, our findings suggest that cardiac fibroblasts may contribute to pathologic hypertrophy in addition to causing fibrosis in primary forms of HCM. TGFβ inhibition may be a useful therapeutic option for patients with HCM due to RASopathies or other etiologies.
Article Information
vol. 130 no. Suppl 2 A19583
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Rebecca Josowitz1;
- Sonia Mulero-Navarro1;
- Christine Falce2;
- Ninette Cohen3;
- Erik M Ullian4;
- Lauren A Weiss5;
- Katherine A Rauen6;
- Eric A Sobie2;
- Bruce D Gelb1
- 1Child Health and Development Institute, Icahn Sch of Medicine at Mount Sinai, New York, NY
- 2Dept of Pharmacology and Systems Therapeutics, Icahn Sch of Medicine at Mount Sinai, New York, NY
- 3Dept of Genetics and Genomics Sciences, Icahn Sch of Medicine at Mount Sinai, New York, NY
- 4Depts of Ophthalmology and Physiology, Univ of California, San Francisco, San Francisco, CA
- 5Dept of Psychiatry, Institute for Human Genetics, Univ of California, San Francisco, San Fransisco, CA
- 6Dept of Pediatrics, UC Davis, Sacramento, CA
Abstract 18890: An MYH7 Mutation Causes Inherited Left Ventricular Noncompaction and Sudden Death in a Large Multigenerational Family
Gina M Morgan, Rebecca Gutmann, Anthony J Klappa, Xiaodong Zhu, Ferhaan Ahmad, Haider Mehdi, Barry London
Circulation. 2014;130:A18890
Abstract
Introduction: Mutations in MYH7, encoding the cardiac β-myosin heavy chain, cause hypertrophic (HCM) and dilated cardiomyopathy (DCM). Recently, several mutations in MYH7 were reported in patients with left ventricular noncompaction (LVNC), a rare cardiomyopathy characterized by increased trabeculation and sudden death (SCD). No large families with inherited LVNC caused by MYH7 mutations have been reported.
Methods: We identified a large family with LVNC and SCD (Pedigree). The proband is a 56 yo woman who presented at age 48 with heart failure, LVNC and a depressed ejection fraction (EF) by echocardiogram (TTE) and cardiac MRI (cMR). Medical records and blood samples were obtained from 20 adult family members, 8 clinically affected (6 LVNC; 2 DCM). DNA was isolated (PureGene), clinical genetic testing for known DCM/LVNC genes was performed on the proband’s affected daughter (GeneDx), and the mutation was identified in family members by direct sequencing. ECGs, TTEs, and cMRs were analyzed to determine PR/QRS/QTc intervals and EF; mutation carriers were compared to non-carriers in the family.
Results: A heterozygous missense mutation in a hydrophobic coil in the myosin head was identified in exon 14 of MYH7 (c.1400 T>C; p.Ile467Thr). This mutation was present in all 8 affected members, in 1 clinically unaffected obligate carrier, and in 1 symptomatic subject who had not undergone cardiac testing (penetrance ≥ 0.80). Three obligate carriers suffered SCD. Compared to noncarriers, the EF of mutation carriers was decreased (42 ± 13%, n=9, vs. 58 ±3%, n=4; p=0.03), the PR was borderline long (171 ± 33 vs. 149 ± 12 ms, n=10 each; p=0.06) and the QTc was prolonged (428 ± 32 vs. 405 ± 18 ms, n=10 each; p=0.05).
Conclusion: The Ile467Thr mutation in MYH7 causes inherited LVNC with high penetrance and SCD in a large family. Future studies are warranted to differentiate the mechanisms leading from this mutation to LVNC as contrasted with those leading from other MYH7mutations to HCM and DCM.
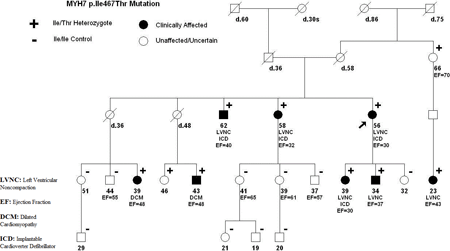
Article Information
vol. 130 no. Suppl 2 A18890
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Gina M Morgan;
- Rebecca Gutmann;
- Anthony J Klappa;
- Xiaodong Zhu;
- Ferhaan Ahmad;
- Haider Mehdi;
- Barry London
- Internal Medicine, Univ of Iowa, Iowa City, IA
Abstract 18613: Improved Adolescent Pre-Athletic Screening Sensitivity and Specificity with Seattle Criteria-interpreted ECGs
Jamie N Colombo, Ricardo Samson, Santiago Valdes, David Sisk, Scott Klewer
Circulation. 2014;130:A18613
Abstract
Introduction: Sudden cardiac arrest (SCA) is a rare but devastating cause of death in children and young adults. While electrocardiogram (ECG) can detect many causes of SCA, ECG are not routinely included in pre-athletic screening (PAS) protocols in the United States due, in part, to a high rate of false positive (FP) interpretations in adolescents. Recently, an expert consensus panel developed refined ECG criteria (Seattle Criteria) to improve the specificity for ECG identification of cardiac conditions associated with SCA.
Methods: We compared standard ECG criteria with Seattle Criteria in the interpretation of ECGs obtained as part of a youth community SCA screening program. Students completed an AHA-endorsed PAS that included a cardiac specific history and physical exam. A 12 lead ECG was obtained and interpreted by a board certified pediatric cardiologist (PDC). Students found to have a positive PAS screen or abnormal standard ECG completed additional work up with a PDC. All ECGs referred for additional evaluation were interpreted by a PDC using Seattle Criteria in a blinded manner.
Results: A total of 1,034 students 11-13 years of age were evaluated. No referrals for additional testing were made based on PAS history alone. There were 72 (7%) ECG abnormalities identified by standard PDC interpretation, while Seattle Criteria identified 21 (2%) ECGs that met criteria for further testing. Formal cardiology evaluation confirmed 4 students at risk for SCA (0.38%); LQTS (n=2), WPW (n=1) and PHTN (n=1); incidental findings isolated RBBB (n=1), MVP (n=1) and PFO (n=1). All students at risk for SCA were identified by both standard pediatric ECG and Seattle Criteria interpretations. Specificity for standard ECG was 93% and for Seattle criteria was 98%.
Conclusions: This study demonstrates that inclusion of ECGs in PAS improves the sensitivity for identifying middle school students at risk for SCA compared to history and exam alone. While standard pediatric ECG interpretation yields a 6.5% incidence of FP, application of Seattle Criteria reduces the incidence of FP ECGs in this age group to 1.6% with no adverse effect on sensitivity. This data supports utilizing Seattle Criteria interpretation for community SCA screening programs that include ECG.
Article Information
vol. 130 no. Suppl 2 A18613
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Pediatrics, Univ of Arizona, Tucson, AZ
- 2Pediatrics, Baylor College of Medicine, Houson, TX
Abstract 17894: Variation in Management of Single Ventricle Patients Undergoing Elective Cardiac Catheterization: a Report from the Congenital Cardiac Catheterization Project on Outcomes (C3PO)
Bryan H Goldstein, Laurie B Armsby, Sara M Trucco, Diego Porras, Joshua J Murphy, Susan R Foerster, Robert H Beekman, Lisa Bergersen
Circulation. 2014;130:A17894
Abstract
Introduction: Variation in management surrounding cardiac catheterization (cath) in patients with single ventricle (SV) heart disease has not been rigorously described and is necessary to inform future quality improvement efforts and evidence-based guideline development.
Objectives: To assess institutional variation in SV cath management.
Methods: Patient and procedural characteristics related to elective SV cath were collected prospectively from 8 centers using a web-based multicenter registry (Congenital Cardiac Catheterization Project on Outcomes). Non-elective cases, those performed <30 days from cardiac surgery, <30 days of age, or in infants receiving prostaglandin therapy were excluded. Cases were stratified by stage of SV palliation (pre-bidirectional cavopulmonary anastomosis (pre-BCPA), pre-Fontan and post-Fontan) for analysis. Institutional variation was assessed.
Results: Between 2/2007 and 6/2010, 1568 elective SV cath procedures met criteria for study inclusion, including 395 pre-BCPA, 611 pre-Fontan and 562 post-Fontan cases. Across institutions, interventional cases comprised 21-63% (median 39%, [IQR 28, 47]) of the pre-BCPA cohort, 50-83% (median 63%, [55, 72]) of the pre-Fontan cohort and 25-91% (median 71%, [54, 82]) of the post-Fontan cohort. Use of positive pressure ventilation varied from 6-100% (median 96%, [56, 100]) in the pre-BCPA cohort, 10-100% (median 99%, [50, 100]) in the pre-Fontan cohort and 0-100% (median 88%, [37, 93]) in the post-Fontan cohort. Adverse event (AE) rates ranged from 0-43% (median 18%, [9, 27]), 0-22% (median 9%, [2, 14]) and 0-19% (median 9%, [5, 17]) in the pre-BCPA, pre-Fontan and post-Fontan cohorts, respectively. Rates of blood transfusion, age at pre-BCPA catheterization, procedure and fluoroscopy times, contrast dose, and key hemodynamic measures (mean PA pressure, cardiac index, ventricular end diastolic pressure) also varied importantly across institutions in the cohorts.
Conclusions: Substantial variation exists in the management surrounding elective cath in SV patients at all stages of palliation. Standardizing management around evidence-based practices may improve outcomes and reduce AE rates for these high-risk children and adults.
Article Information
vol. 130 no. Suppl 2 A17894
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Bryan H Goldstein1;
- Laurie B Armsby2;
- Sara M Trucco3;
- Diego Porras4;
- Joshua J Murphy5;
- Susan R Foerster6;
- Robert H Beekman1;
- Lisa Bergersen4
- 1The Heart Institute, Cincinnati Children’s Hosp Med Cntr, Cincinnati, OH
- 2Pediatric Cardiology, Oregon Health Sciences Univ, Portland, OR
- 3Heart Institute, Children’s Hosp of Pittsburgh of UPMC, Pittsburgh, PA
- 4Cardiology, Boston Children’s Hosp, Boston, MA
- 5Pediatric Cardiology, St. Louis Children’s Hosp, St. Louis, MO
- 6Pediatric Cardiology, Children’s Hosp of Wisconsin, Milwaukee, WI
Abstract 16375: Low Nasal Nitric Oxide as a Prognosticator of Failing Single Ventricle Physiology in Congenital Heart Disease Patients?
Cyrus Yau, Maliha Zahid, Omar Khalifa, William Devine, Linda Leatherbury, Peter Wearden, Cecilia Lo
Circulation. 2014;130:A16375
Abstract
Introduction: Pulmonary arterial hypertension (PAH) is a major complication in patients with congenital heart disease (CHD). The mechanism of PAH in CHD is multi-factorial and includes increased pulmonary blood flow/pressure, shear stress to pulmonary arteries, and vasoconstriction, proliferation, and thrombosis to the pulmonary vascular bed. PAH patients are treated with medications to increase nitric oxide (NO) levels, as reduction of endothelial-derived NO is thought to contribute to PAH pathogenesis. Interestingly, we recently showed some CHD patients can have very low nasal nitric oxide (nNO), prompting us to investigate the hypothesis that CHD patients with low nNO may have increased risk for PAH.
Methods: Retrospective chart review of cardiac catheterization was performed and pulmonary arterial hemodynamic data were analyzed in 85 CHD patients. Nasal NO measurements were obtained, and patients with low nNO were identified using nNO cutoff values used for diagnosis of primary ciliary dyskinesia (PCD), a sinopulmonary disease associated with very low nNO.
Results: No significant correlation was found between low nNO and mean pulmonary artery pressures, indexed PVR, or pulmonary blood flow in CHD patients. However, CHD patients with failing Fontan/bidirectional Glenn (BDG) circulations and functional BDG had significantly lower nNO as compared to CHD patients with functioning Fontan circulations (p < 0.0001). Furthermore, CHD patients with single ventricle physiology were 14 times more likely to fail if they have extremely low nNO levels below the PCD cut off values (OR 14.8, p = 0.006). In several patients, nNO measurements before and after heart transplantation showed persistence of the nNO deficiency after transplantation, suggesting an intrinsic defect in nNO production unrelated to post-surgical alterations in pulmonary blood flow.
Conclusions: In CHD patients, there was no correlation between low nNO and PAH, but instead, a highly significant correlation was observed between low nNO and failing Fontan/BDG circulations. These novel findings suggest further studies are warranted to investigate whether low nNO may be a prognosticator of poor outcome in Fontan subjects and thus may guide clinical care of such high-risk CHD patients.
Article Information
vol. 130 no. Suppl 2 A16375
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Cyrus Yau1;
- Maliha Zahid2;
- Omar Khalifa2;
- William Devine3;
- Linda Leatherbury4;
- Peter Wearden5;
- Cecilia Lo2
- 1Pediatrics, Div of Pediatric Cardiology, Children’s Hosp of Pittsburgh, Pittsburgh, PA
- 2Developmental Biology, Univ of Pittsburgh Sch of Medicine, Pittsburgh, PA
- 3Pediatric Pathology, Univ of Pittsburgh Sch of Medicine, Pittsburgh, PA
- 4Pediatric Cardiology, Children’s National Med Cntr, Washington, DC
- 5Div of Pediatric Cardiothoracic Surgery, Children’s Hosp of Pittsburgh, Pittsburgh, PA
Abstract 12791: Acute Volume Loading During Invasive Testing Identifies Fontan Survivors With Occult Diastolic Dysfunction
Konstantin Averin, Russel Hirsch, Michael Seckeler, Robert H Beekman, Bryan H Goldstein
Circulation. 2014;130:A12791
Abstract
Introduction: Invasive hemodynamic assessment of the Fontan patient is often unrevealing, even in the presence of clinical symptoms. Diastolic dysfunction (DD), a key driver of long-term clinical outcomes, may frequently be concealed during standard hemodynamic evaluation.
Hypothesis: Volume loading during invasive assessment may identify Fontan patients with occult DD.
Methods: Single center record review of cardiac catheterizations with volume loading in Fontan patients from 2012 – 2014. Patients with baseline DD, defined as a ventricular end diastolic pressure of ≥ 15 mmHg, were excluded. Following acquisition of baseline data, a 15 ml/kg normal saline intravenous bolus was administered rapidly. After a 5 min equilibration phase, hemodynamics were reassessed. Comparisons were made using paired Student’s t-test. Risk factors for occult DD were assessed using Wilcoxon rank-sum test.
Results: Twenty-three Fontan patients (48% female, 74% left ventricular morphology) were included. Median age was 17.7 (IQR 7.5, 21.4) years and mean duration of Fontan circulation was 12.9 ± 9.1 years. Hemodynamic data are shown in Table 1. Volume loading revealed occult DD in 8 (35%) patients (Fig 1). Age (21.3 vs. 11.8 years, p=0.05) and duration of Fontan circulation (21.1 vs. 10.6 years, p=0.04) were associated with occult DD, whereas ventricular morphology was not. There were no adverse events related to volume loading.
Conclusions: Occult DD is common in Fontan patients, and may be identified with volume loading during invasive hemodynamic assessment. Older age and longer duration of Fontan circulation were associated with a greater likelihood of occult DD.
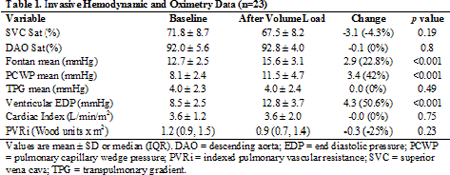
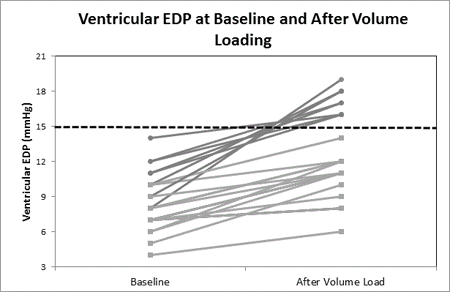
Article Information
vol. 130 no. Suppl 2 A12791
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1The Heart Institute, Cincinnati Children’s Hosp, Cincinnati, OH
- 2Pediatric Cardiology, The Univ of Arizona, Tucson, AZ
Abstract 11683: Recurrence of Atrial Arrhythmias Following Ablation in Adults With Congenital Heart Disease: New Substrate Formation or Late Procedural Failure?
Jacqueline Shuplock, Gregory Barker, Andrew Radbill, Prince Kannankeril, Frank A Fish
Circulation. 2014;130:A11683
Abstract
Introduction: Using current mapping and ablation tools, high acute success rates can be achieved in repaired adult congenital heart disease (CHD) patients with atrial arrhythmias, but recurrences are common. We examined the nature of recurrence in a cohort of repaired adult CHD patients.
Hypothesis: We tested the hypothesis that recurrences after ablation in repaired CHD patients represent the development of new substrates.
Methods: A retrospective chart review was performed on all repaired adult CHD patients undergoing ablation for atrial arrhythmias, excluding atrial fibrillation (AF), at our institution from Jan. 2003-Nov. 2013. CHD lesions were grouped as tetralogy-type, atrial switch, single ventricle, and other. Latest follow-up data were obtained on all patients. Acute procedural success was defined as ablation of all targeted circuits. All recurrences warranting clinical intervention were noted.
Results: A total of 118 patients underwent 157 ablation procedures, and 253 tachycardias were targeted. Of these, 99 utilized the cavo-tricuspid isthmus (CTI), 88 were non-isthmus-dependent scar-related macro-reentry, 55 were focal tachycardias, and 13 were atrioventricular reciprocating tachycardias. Acute procedural success was obtained in 147/157 (94%) procedures and 237/253 (94%) targeted arrhythmias. Recurrence was observed after 78 (50%) procedures after a median time of 6 months. Of those undergoing repeat ablations, new substrates were found to be the cause of recurrence in 18 (23%), while recurrence of the original substrate was documented in 20 (26%). AF accounted for 9 (12%) of the recurrences. By lesion type, recurrence was most common among single ventricle patients (81% vs. all others 42%, p <0.001).By substrate, recurrence was lowest with CTI-dependent circuits (35% vs. all others 56%, p=0.016).
Conclusions: Despite high acute procedural success rates, patients with repaired CHD had frequent recurrence of atrial arrhythmias. These represented both new substrates, including the development of AF, and recurrence of previously-ablated substrates with a similar frequency. Repeat ablation may be warranted in those with recurrences as a new target for ablation may be identified.
Article Information
vol. 130 no. Suppl 2 A11683
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Jacqueline Shuplock;
- Gregory Barker;
- Andrew Radbill;
- Prince Kannankeril;
- Frank A Fish
- Pediatric Cardiology, Vanderbilt Univ Med Cntr, Nashville, TN
Abstract 20047: Nasa Model of “Threat and Error” in Paediatric Cardiac Surgery: Death Typically Results From Cycles of Error That Originate in the Operating Room and Are Amplified by Additional Error in Intensive Care
Edward Hickey, Yaroslavna Nosikova, Eric Pham-Hung, Michael Gritti, Travis Wilder, Sara Hussain, Christopher A Caldarone, Steven Schwartz, Andrew Redington, Glen S Van Arsdell
Circulation. 2014;130:A20047
Abstract
Introduction: We introduced the NASA “threat and error model” to our surgical unit; all admissions are considered “flights”, which should pass through stepwise de-escalations in risk.
Hypothesis: Errors significantly influence risk de-escalation and contribute to poor outcomes.
Methods: Patient flights (524) were tracked real-time for threats, errors and unintended states (figure). Expected risk de-escalation was: wean from mechanical support, sternal closure, extubation, ICU discharge and discharge home. Data were accrued via performance personnel, bedside data, reporting mechanisms and staff interviews. Infographics of flights were openly discussed weekly.
Results: In 12% (64/524) of flights, the child failed to de-escalate sequentially through expected risk levels; unintended increments instead occurred. Failed de-escalations were highly associated with errors (426; 257 flights), however seemingly benign (P<.0001). Errors with clinical consequence (263; 173 flights) had 29% rate of failed de-escalations vs 4% (P<.0001).
The most dangerous errors were “apical” errors typically (84%) occurring in the OR which led to cycles of propagating unintended states (n=110): these had 43% (47/110) rate of failed de-escalation (vs 4%, P<.0001). Apical errors were triggered by identifiable threats in 25% (28/110) (usually – 75% – morphology/ comorbidities); 75% were instead “unforced” errors. Cycles of unintended state were often (46%) amplified by additional (up to 7) errors in ICU that would worsen clinical deviation. Overall, failed de-escalations in risk were extremely closely linked to brain injury (N=13; P<.0001), or death (N=7; P<.0001).
Conclusions: Deaths and brain injury almost always occur from propagating error cycles that originate in the OR and are often amplified by additional ICU errors. Improvements in threat management, error detection/rescue and vigilance at times of failed de-escalation will translate into improved outcomes.
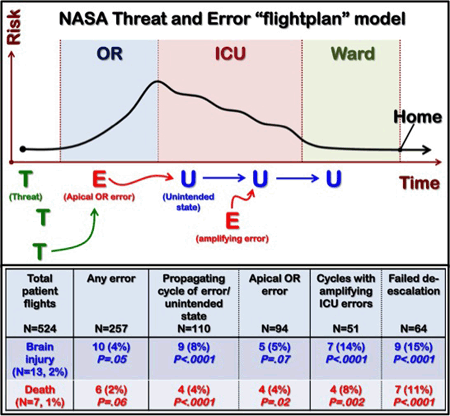
Article Information
vol. 130 no. Suppl 2 A20047
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Edward Hickey1;
- Yaroslavna Nosikova1;
- Eric Pham-Hung1;
- Michael Gritti1;
- Travis Wilder1;
- Sara Hussain1;
- Christopher A Caldarone1;
- Steven Schwartz2;
- Andrew Redington3;
- Glen S Van Arsdell1
- 1Cardiovascular Surgery, The Hosp for Sick Children, Toronto, Canada
- 2Critical Care, The Hosp for Sick Children, Toronto, Canada
- 3Cardiology, The Hosp for Sick Children, Toronto, Canada
Abstract 18082: Obstructive Sleep Apnea Independently Mediates Cardio-Metabolic Risk in Obese Adolescents
Indra Narang, Brian W McCrindle, Cedric Manlhiot, Catherine Birken, Jill Hamilton
Circulation. 2014;130:A18082
Abstract
Introduction: Obstructive sleep apnea (OSA) is characterized by snoring, recurrent obstruction of the upper airway and intermittent desaturations. OSA complicates obesity in 30% of adolescents. Chronic untreated OSA in adults is associated with increased cardiometabolic (CM) risk but limited data in obese adolescents are conflicting and suggest that body mass index (BMI) primarily confers CM risk rather than OSA.
Objective: To evaluate the impact of OSA on CM risk in obese adolescents. We hypothesized that OSA independently mediates CM risk in obese adolescents.
Methods: This was a cross-sectional, prospective study where obese children and adolescents, aged 8-18 years without a history of OSA were consecutively recruited. After an overnight fast, participants had standardized measurements of height, weight, waist circumference and blood pressure. BMI z scores and waist to height ratio (WHtR) were calculated. Fasting blood samples for lipid profile, insulin, glucose, high sensitivity C-reactive protein (CRP) were obtained. All participants underwent an overnight polysomnography. An obstructive apnea-hypopnea index (OAHI) of ≤5 events/hour was used to define OSA.
Results: 105 obese participants (45% males) were recruited and 27/105 (26%) had OSA. In the OSA group versus the no-OSA group, the mean age (±SD) in years was 15.1±2.5 and 14.7±2.5 respectively (p=0.45). The mean BMI (SD) z scores and WHtR (SD) were significantly higher in the OSA group compared to the no-OSA group (2.66±0.54 vs 2.36±0.49, respectively, p=0.01 and 0.75±0.16 vs 0.66±0.12 respectively, p=0.01). After adjusting for age, gender and WHtR in multivariable linear regression model, OAHI ≥5 was significantly associated with greater CM risk, specifically, higher fasting insulin (EST (SE): +243 (93) pmol/L, p=0.01), higher HOMA-IR: (7.7 (3.1), p=0.01), lower high density lipoprotein (-0.60 (0.23) mmol/L, p=0.01), higher CRP (+16 (5) mg/L, p=0.001) and higher systolic and diastolic BP z scores (+2.9 (0.9) z systolic, +2.2 (0.6) z diastolic, p=0.001 for both).
Conclusion: OSA in the context of obesity may further potentiate CM risk in adolescents. Early screening and targeted therapeutic interventions for OSA should be optimized in obese youth to minimize long-term CM risk.
Article Information
vol. 130 no. Suppl 2 A18082
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Indra Narang;
- Brian W McCrindle;
- Cedric Manlhiot;
- Catherine Birken;
- Jill Hamilton
- Labatt Family Heart Cntr, The Hosp for Sick Children, Toronto, Canada
Abstract 16769: Race to the Top: Are Children With Congenital Heart Disease Off to a False Start?
Matthew Oster, Alexandra Ehrlich, William T Mahle, Jessica Knight, Bryan Williams
Circulation. 2014;130:A16769
Abstract
Background: Clinical trials and registries have found that children with congenital heart disease (CHD) have poorer neurodevelopment than their unaffected peers. Recent emphasis on mandatory school-based assessment, including as part of the U.S. Department of Education Race to the Top program, provides a unique opportunity to assess how such impairment impacts school performance. The purpose of our study was to evaluate the association of CHD with a) performance on standardized tests and b) the need for special education services.
Methods: We performed a retrospective cohort study comparing school performance for 438 children born 2002-2003 with a history of surgery for CHD at our institution vs. that of children in our state without CHD. Our two primary outcome measures were a) the % of children meeting standards on the various portions of the 3rd grade Criterion Referenced Competency Test and b) the % of children receiving an individualized education plan for special education services in 2008-2011.
Results: Children with CHD were more likely not to meet standards in math or social studies, but were more likely to meet standards for reading. There were no significant differences in English or science. (Figure) Among those with CHD, presence of a non-cardiac abnormality was a risk factor for not meeting standards (p<0.01) but surgical risk category was not. In all years, children with CHD were significantly more likely to receive special education services (range 28% – 45% per year) as compared to other children (range 11%-12% per year, p<0.0001).
Conclusions: CHD is an important risk factor for poor performance on the math and social studies portions of standardized testing, but increased CHD surgical risk does not translate to poorer school performance. Students with CHD are receiving special education services at increased proportions as compared to their peers, a fact that may explain improved reading performance in those with CHD as compared to their peers.
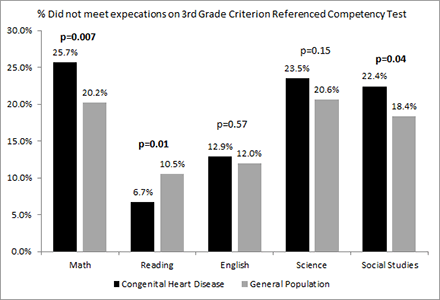
Article Information
vol. 130 no. Suppl 2 A16769
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Sibley Heart Cntr Cardiology, Children’s Healthcare of Atlanta, Atlanta, GA
- 2Sch of Public Health, Emory Univ, Atlanta, GA
- 3Sch of Nursing, Emory Univ, Atlanta, GA
Abstract 15499: Children After Fontan have the Strength, Body Composition, and Daily Moderate-to-Vigorous Physical Activity Required for Successful Participation with Peers
Patricia E Longmuir, Mary Corey, Guy Faulkner, Jennifer L Russell, Brian W McCrindle
Circulation. 2014;130:A15499
Abstract
Introduction: This cross-sectional study evaluated the healthy, active lifestyle capacity (daily physical activity, strength, flexibility, body composition) of children after Fontan, which was hypothesized to be lower than healthy peers.
Methods: Participants (n=64, 25 female) were 9 ± 2 years of age. Fontan completion occurred at 3 ± 1 years of age. Canadian Health Measures Survey protocols assessed aerobic endurance (walking up/down steps at set pace), strength (handgrip dynamometry), flexibility (sit and reach), body composition (body mass index) and daily moderate-to-vigorous physical activity (7-day accelerometry). Participant versus published norm differences were evaluated with t-tests. Linear regression evaluated associations with age/gender/demographic factors.
Results: Children after Fontan had strength scores similar (mean difference 1 kg) to their peers, were less likely to be obese (mean difference of body mass index = 1.1 ± 2.5, p=.001) and performed 50 minutes of moderate-to-vigorous activity per day. Estimated maximal aerobic endurance (mean difference = 21 ± 3 ml/kg/min or 61% of expected) and flexibility (mean difference = 9 ± 8 cm or 64% of expected) were lower than peers(p<.001). Participants performed fewer minutes of daily activity (mean difference from normal 12 ± 17 minutes/day, p<.001), but almost all (60/63) demonstrated the capacity for at least 20 minutes per day. Daily activity was higher with Fontan completion at a younger age (4 ± 2 mins/year) and for those taking antithrombotic medication (7 ± 18 and 22 ± 17 fewer minutes/day for those taking/not taking antithrombotics, respectively).
Conclusions: Children after Fontan demonstrate the capacity to successfully perform the daily physical activity associated with optimal health. They have similar levels of strength and good body composition. We recommend that children after Fontan be counselled to expect that they can successfully participate in physically active peer play.
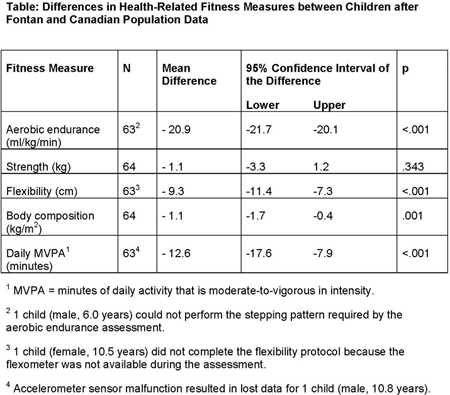
Article Information
vol. 130 no. Suppl 2 A15499
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Healthy Active Living and Obesity Rsch Group, Children’s Hosp of Eastern Ontario Rsch Institute, Ottawa, Canada
- 2Child Health Evaluative Sciences, Hosp for Sick Children, Toronto, Canada
- 3Faculty of Kinesiology and Physical Education, Univ of Toronto, Toronto, Canada
- 4Labatt Family Heart Cntr, Hosp for Sick Children, Toronto, Canada
Abstract 15517: Receipt of Special Education Services Among Children with Congenital Heart Defects in Atlanta, Georgia
Tiffany Colarusso, Autry Andrew, Hilda Razzaghi, Coleen Boyle, William Mahle, Kim Van Naarden Braun, Adolfo Correa
Circulation. 2014;130:A15517
Abstract
BACKGROUND: Population-based information on special education service needs among children with congenital heart defects (CHDs) is limited. We investigated the prevalence of receipt of special education services among children with CHDs.
METHODS: Children born from 1982-2004 in metropolitan Atlanta with CHDs (n=3,744) were identified from a population-based birth defect surveillance program, and contemporaneous children born without major birth defects (n=860,715) were identified from birth certificates. Cohorts were linked to special education files for 1992-2012 school years to identify receipt of special education services. Children with CHDs and non-cardiac defects or genetic syndromes were excluded; children with isolated CHDs were classified by presence or absence of critical CHDs (i.e., defects needing intervention in the first year of life). We evaluated prevalence of special education services and calculated prevalence ratios (PRs) using children without birth defects as reference.
RESULTS: Children with CHDs were 50% more likely than those without birth defects to use special education services (PR 1.5; 95% Confidence interval 1.4-1.7). Similar to children without birth defects, the most common eligibility among children with CHDs was speech/language impairment. Compared to children without birth defects, the prevalence of several special education eligibilities were significantly higher among children with all CHDs: any intellectual disability (PR 3.8), sensory impairment (PR 3.0), other health impairment (PR 2.8), significant developmental delay (PR 1.9), orthopedic impairment (PR 1.9), and specific learning disability (PR 1.4). For most special education eligibilities, there was no significant variation in the elevated prevalence ratios by presence or absence of critical CHDs.
CONCLUSIONS: Children with many types of CHDs received special education services more often than children without birth defects. These findings highlight important resource needs for children with CHDs. Furthermore, they suggest that recommendations to perform developmental screening only on children with select CHDs may miss or delay identification of children with other CHDs, who may also need special education services.
Article Information
vol. 130 no. Suppl 2 A15517
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Tiffany Colarusso1;
- Autry Andrew1;
- Hilda Razzaghi1;
- Coleen Boyle1;
- William Mahle2;
- Kim Van Naarden Braun1;
- Adolfo Correa3
- 1National Cntr on Birth Defects and Developmental Disabilities, CDC, Atlanta, GA
- 2Sibley Heart Cntr, Children’s Healthcare of Atlanta, Atlanta, GA
- 3Dept of Medicine and Pediatrics, Univ of Mississippi Med Cntr, Jackson, MS
Abstract 11740: Fetal and Neonatal Markers of Neurodevelopmental Outcome in Congenital Heart Disease
Ismee A Williams, Howard Andrews, Michael M Myers, William Fifer
Circulation. 2014;130:A11740
Abstract
Objectives: Children with congenital heart disease (CHD) are at risk for abnormal neurodevelopment (ND). We evaluated associations between fetal Doppler and biometry measures, neonatal electroencephalogram (EEG) and 18-month ND.
Methods: Fetuses with hypoplastic left heart syndrome (HLHS), transposition of the great arteries (TGA), and tetralogy of Fallot (TOF) had middle cerebral (MCA) and umbilical artery (UA) Doppler velocities, as well as biometry such as head (HC) and abdominal circumference (AC), prospectively recorded at 20-25 (F1), 26-32 (F2), and 33-39 (F3) wks gestational age (GA). Pulsatility indices (PI) with GA-derived z-scores and cerebral-to-placental resistance (CPR) ratios were calculated. Neonatal high-density EEG was preformed preoperatively and the Bayley Scales of Infant Development-III were assessed at 18-months. Factor analysis was used to reduce the number of EEG predictors used in regression analysis.
Results: Among 56 CHD fetuses (N=19 HLHS, N=16 TGA, N=21 TOF) who underwent preoperative EEG, ND scores are available for 33 to date. Cardiac subtype was highly associated with EEG and was considered in all models. Cognition scores were predicted by CPR< 1 ever (B=-15.7, P=0.002) and HC/AC at F2 (B=-130, P=0.013, R2=0.42). Language scores were predicted by UA PI z-score at F1 (B=-9.6, P=0.005, R2=0.27). Motor scores were predicted by UA PI z-score at F1 (B=-3.9, P=0.085), HLHS (B=-15, P<0.001), EFW%ile (B=0.374, P=0.007), and delta band right parietal and right temporal log power in active sleep (B=3.9, P=0.045, R2=0.61).
Conclusion: Lower umbilical artery pulsatility at 20-25 wks GA was associated with higher 18-month Language and Motor scores. A diagnosis of HLHS predicted poorer Motor scores. Increased EEG power in the parietal and temporal region of the right brain predicted higher Motor scores. A larger abdomen relative to head at 26-32 wks was associated with improved cognition while diminished cerebrovascular compared with placental resistance predicted poorer cognition, similar to what has been observed in the growth restricted fetus. Further investigation is needed to confirm these hypothesis-generating findings.
Article Information
vol. 130 no. Suppl 2 A11740
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Pediatrics, Div of Pediatric Cardiology, Morgan Stanley Children’s Hosp of NewYork-Presbyterian, Columbia Univ Med Cntr, New York, NY
- 2Biostatistics, Mailman Sch of Public Health, Columbia Univ, New York, NY
- 3Developmental Neuroscience, New York State Psychiatric Institute, New York, NY
Abstract 11083: Pulse Oximetry Overestimates Arterial Oxy-Hemoglobin in Neonates With Known Critical Congenital Heart Disease (CCHD) – Implications for Newborn Screening
Daniel P Murphy, John P Cleary
Circulation. 2014;130:A11083
Abstract
Background: Pulse oximetry is a key part of the clinical management of CCHD and more recently, in 2011, the United States adopted recommendations to use pulse oximetry to screen for critical congenital heart disease (CCHD). CCHD is estimated to occur in about 0.25% of live births. Recently, the accuracy of the pulse oximetry in PICU patients has been questioned, especially in hypoxemic patients.
Objective: Compare SpO2 and oxy-Hgb values in patients with known CCHD to evaluate the precision of non-invasive measurements of arterial saturations in CCHD management and screening.
Methods: Single-Center Retrospective Study. Inclusion criteria: AHA defined CCHD and a post ductal arterial blood gas in first 72 hours of life. 71 patients with 466 measurements of SpO2 (Masimo Inc., Irvine, CA) were analyzed to determine correlation with arterial oxy-hemoglobin (Siemens, Erlangen, GE). Paired T-Test and ANOVA utilized to compare data points (SPSS v21).
Results: SpO2 overestimates arterial oxy-Hgb by a mean difference of 4.6% in all patients with CCHD. The mean variance was statistically significant with all groups including those with SpO2 >95% (5.6%), SpO2 90-94% (5.7%), SpO2 80-89% (4.6%), SpO2 70-79% (4.5%), and SpO2 >=6% in 46% of all paired measurements. Only 4% of SpO2 measurements underestimated oxy-Hgb by >3%. Hour of life was not statistically significant.
Conclusion: This data set raises concern that present pulse oximeters might have a meaningful false negative rate in CCHD screening. In infants less than 72 hours old with CCHD, pulse oximetry was found to significantly overestimate oxy-Hgb with a mean variance of 4.6%. For example, Truncus Arteriosis and Hypoplastic Left Heart Syndrome both had average SpO2 readings of 94% in the first 72 hours of life while OxyHgb measured between 87.6%-88.8%. Clinical decisions are frequently made based on non-invasive pulse oximetry, which may lead to inaccurate predictions of qp/qs and inappropriate escalation in therapy. These results raise concern that present pulse oximetry algorithms may be sub-optimal in CCHD newborns.
Article Information
vol. 130 no. Suppl 2 A11083
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Div of Neonatology, Harbor UCLA / Children’s Hosp Orange County, Torrance, CA
- 2Div of Neonatology, Children’s Hosp Orange County, Orange, CA
Abstract 11256: Handheld Echocardiography Improves Detection of Rheumatic Heart Disease in Ugandan Schoolchildren Compared to Auscultation
Justin Godown, Jimmy C Lu, Andrea Beaton, Craig Sable, Grace Mirembe, Richard Sanya, Twalib Aliku, Sunkyung Yu, Gregory J Ensing
Circulation. 2014;130:A11256
Abstract
Background: Rheumatic heart disease (RHD) remains a major public health concern in developing countries and routine screening has the potential to improve outcomes. Standard portable echocardiography (SPE) is far more sensitive than auscultation for the detection of RHD, but remains cost-prohibitive in resource-limited settings. With its lower cost, handheld echocardiography (HHE) has the potential to fill this void. The purpose of this study was to assess the incremental value of HHE over auscultation to identify RHD, as compared to SPE.
Methods: Over 1 week, children at 5 schools in Gulu, Uganda underwent focused SPE (parasternal and apical views). Any child with mitral or aortic regurgitation or stenosis, plus a randomly selected group of children with normal SPE findings underwent HHE by echocardiographers and auscultation by a local physician. SPE and HHE studies were interpreted blindly using the 2012 World Heart Federation criteria by 6 experienced cardiologists. A second reader confirmed any study with borderline or definite RHD, with discrepancies resolved by a third reader. Sensitivity and specificity of HHE and auscultation for the detection of any RHD, definite RHD, and pathologic mitral or aortic regurgitation were calculated using SPE as the gold standard.
Results: Of 4,773 children who underwent screening with SPE, a subgroup of 1,317 children (46 % male, 10.8 ± 2.6 years of age) underwent HHE and auscultation. Auscultation had uniformly poor sensitivity to detect RHD or valve disease. Sensitivity was significantly improved using HHE compared to auscultation for the detection of definite RHD (97.8% vs 22.2%), borderline or definite RHD (78.4% vs 16.4%), and pathologic aortic insufficiency (81.8% vs 13.6%) (Table).
Conclusions: Auscultation is a poor screening test for the detection of RHD. HHE significantly improves detection of RHD and may be a useful adjunct to or replacement of auscultation in resource-limited settings.
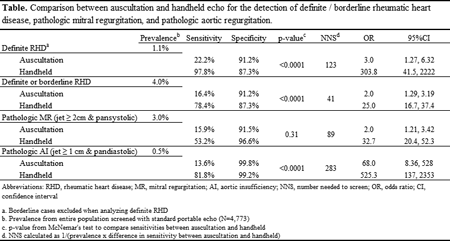
Article Information
vol. 130 no. Suppl 2 A11256
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Justin Godown1;
- Jimmy C Lu1;
- Andrea Beaton2;
- Craig Sable2;
- Grace Mirembe3;
- Richard Sanya4;
- Twalib Aliku5;
- Sunkyung Yu1;
- Gregory J Ensing1
- 1Pediatric Cardiology, Univ of Michigan, Ann Arbor, MI
- 2Pediatric Cardiology, Children’s National Med Cntr, Washington, DC
- 3Joint Clinical Rsch Cntr, Joint Clinical Rsch Cntr, Kampala, Uganda
- 4Medicine, Gulu Univ, Kampala, Uganda
- 5Uganda Heart Institute, Gulu Univ, Kampala, Uganda
Abstract 19908: 5-oxoprolinase: a Novel Cardiac Mediator of the Oxidative Stress Response in the Failing Heart
Atze van der Pol, Jasper Tromp, Martijn F Hoes, Ibrahim J Domian, Wiek H van Gilst, Herman Silljé, Rudolf A de Boer, Peter V van der Meer
Circulation. 2014;130:A19908
Abstract
Introduction: A hallmark of Heart Failure (HF) is the re-emergence of the fetal gene program. The aim of this study was to identify novel genes associated with the fetal gene program in HF, by utilizing a mouse embryonic stem (mES) cell based micro-array.
Methods/Results: The micro-array analysis was performed on mES cells, early and late mES-derived cardiac progenitors, and mouse neonatal right ventricular tissue. As expected, our screen effectively identified established fetal genes, including alpha smooth muscle actin. We further uncovered several novel genes behaving like fetal genes. The top 5 hits were validated in an in vivo pressure overload HF model. 5-oxoprolinase (OPLAH) was found to be the most significantly differentially expressed gene in the HF model. In a human organ panel, OPLAH gene expression was highest in cardiac tissue, and found to be expressed mainly in the cytosol of cardiomyocytes. Additionally OPLAH protein levels were reduced by 50-70% in both pressure overload and ischemia induced HF (p=0.001 and p=0.009, respectively). OPLAH is an ATP-hydrolyzing enzyme involved in the γ-Glutamyl cycle, responsible for the conversion of the metabolite 5-oxoproline into glutamate. To study OPLAH function we used siRNA and/or over-expression by means of viral transfection of neonatal rat ventricular myocytes. In vitro silencing of OPLAH resulted in an increased susceptibility towards oxidative stress induced apoptosis (40% increase in cleaved caspase-3 positive cells, p=0.02). Furthermore, exogenous administration of 5-oxoproline increased apoptosis in cardiomyocytes (40% increase in cleaved caspase-3 positive cells, p=0.04).
Conclusions: We identified OPLAH as a novel enzyme associated with HF, behaving like fetal genes, which is involved in the innate protection against oxidative stress, by scavenging excess 5-oxoproline.
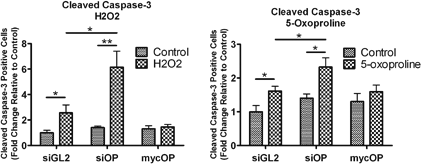
Article Information
vol. 130 no. Suppl 2 A19908
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Atze van der Pol1;
- Jasper Tromp1;
- Martijn F Hoes1;
- Ibrahim J Domian2;
- Wiek H van Gilst1;
- Herman Silljé1;
- Rudolf A de Boer1;
- Peter V van der Meer1
- 1Experimental Cardiology, UMCG, Groningen, Netherlands
- 2Richard B. Simches Rsch Cntr, Massachusetts General Hosp, Boston, MA
Abstract 18834: Genetic Ablation of S-nitrosoglutathione Reductase (GSNOR) in Mice Enhances Proliferative Expansion of Adult Heart Progenitors and Myocytes Post Myocardial Infarction
Konstantinos E Hatzistergos, Ellena C Paulino, Raul A Dulce, Lauro M Takeuchi, Shathiyah Kulandavelu, Wayne Balkan, Rosemeire M Kanashiro-Takeuchi, Joshua M Hare
Circulation. 2014;130:A18834
Abstract
Introduction: The molecular pathways underlying the proliferative activity of adult cardiac myocytes and stem/progenitors in response to heart damage remain elusive. Since perturbing nitroso-redox balance in mice by genetic deletion of GSNOR (GSNOR-/-) confers resilience to experimental myocardial infarction (MI), we investigated whether GSNOR knockout influences the proliferative activity of the post-MI heart.
Hypothesis: Knockout of GSNOR in mice enhances the proliferative expansion of CMs and cKit+ cardiac stem cells (CSCs) in response to MI.
Methods: Wild-type (WT) and GSNOR-/-mice (n=5/ group) underwent experimental MI. To assess proliferative activity, animals received intraperitoneal injections of 5-bromodeoxyuridine (BrdU) at selected time-points during the first 2 weeks post-MI. Immunnohistochemical evaluation was performed 1 month post-MI.
Results: Confocal immunofluorescence revealed that GSNOR-/- hearts exhibited higher rates of BrdU incorporation in CSCs after MI (10.9%±4.99 of WT CSCs compared to 15.9%±3% of GSNOR-/- CSCs, p=0.02). Similarly, there were ~3-fold more BrdU+/Tropomyosin+cardiomyocytes in the infarct zone of GSNOR-/- mice compared to WT (p<0.05). Immunohistochemical evaluation of cardiac troponin-T+ cardiomyocytes co-expressing the mitotic marker ser-10 phosphorylated histone H3 (H3P) showed further that cardiomyocyte mitosis was 2.4-fold greater in GSNOR-/- compared to WT mice (p<0.05), whereas the presence of aurora-b kinase in the cleavage furrow of GSNOR-/- cardiomyocytes substantiated their competence for mitosis after MI. The rate of cardiomyocyte apoptosis after MI, was not different between GSNOR-/- and WT mice, as shown by activated cleaved caspase-3 immunofluorescence.
Conclusions: Collectively, our findings suggest that protein S-nitrosothiol turnover by GSNOR regulates proliferation of cardiomyocytes and CSCs in the adult heart in response to damage. These findings have therapeutic implications for the treatment of heart disease since they reveal novel pathways by which nitroso-redox balance influences cardiac repair.
Article Information
vol. 130 no. Suppl 2 A18834
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Konstantinos E Hatzistergos;
- Ellena C Paulino;
- Raul A Dulce;
- Lauro M Takeuchi;
- Shathiyah Kulandavelu;
- Wayne Balkan;
- Rosemeire M Kanashiro-Takeuchi;
- Joshua M Hare
- Interdisciplinary Stem Cell Institute, Univ of Miami, ISCI, Miami, FL
Abstract 17037: CTCF Regulates Fetal Genes and is Inversely Correlated with Cardiac Muscle Size Across 100 Mouse Strains
Manuel Rosa-Garrido, Christoph Rau, Elizabeth Soehalim, Jessica Wang, Aldons J Lusis, Yibin Wang, Thomas M Vondriska
Circulation. 2014;130:A17037
Abstract
Heart failure is accompanied by abnormal gene expression, however the global chromatin structural mechanisms responsible for transcriptional changes are unknown. We hypothesized that specific chromatin structural proteins orchestrate gene expression in the normal and diseased myocyte. The conserved zinc finger protein CTCF has been shown in diseases such as cancer to regulate transcription, enhancer function, and maintenance of 3D chromatin structure. The objective of this study was to investigate the role of CTCF in cardiac growth and pathology.
We conducted an unbiased systems genomics analysis-in which ~100 strains of mice were treated with isoproterenol (3 wk) followed by transcriptome and phenotype analyse-to investigate the genetic architecture of heart failure. This analysis revealed a strong negative correlation between CTCF mRNA expression and cardiac hypertrophy as measured by heart weight (r2=0.076, p=9.7E-9), left ventricular mass (r2=0.053, p=9.8E-8) and left ventricular internal diameter in diastole (r2=0.033, p=1.6E-5; association with ejection fraction was not significant, r2=0.007, p=0.63). We observed an 80% down-regulation of CTCF at the mRNA level, and 60% at the protein level, in neonatal rat ventricular myocytes (NRVMs) 48h following treatment with either isoproterenol (1umol) or phenylephrine (10umol). RT-PCR in agonist-treated NRVMs confirmed activation of the fetal gene program. However, siRNA mediated CTCF knockdown resulted in the opposite scenario, wherein SERCA and alpha-MHC were up-regulated while ANF and beta-MHC were down-regulated. The effect of CTCF KD on fetal gene expression is still observed when cells are simultaneously treated with hypertrophic agonists, suggesting that loss of CTCF partially prevents activation of these genes following acute hypertrophic stress. Lastly, CTCF KD induced cell death in NRVMs, implying that CTCF is an essential protein for myocyte survival. These data indicate that CTCF is involved in preservation of normal gene expression patterns in the healthy heart and its levels are genetically correlated with cardiac muscle size.
Article Information
vol. 130 no. Suppl 2 A17037
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Manuel Rosa-Garrido1;
- Christoph Rau1;
- Elizabeth Soehalim1;
- Jessica Wang2;
- Aldons J Lusis3;
- Yibin Wang4;
- Thomas M Vondriska4
- 1Anesthesiology, UCLA, Los Angeles, CA
- 2Medicine, UCLA, Los Angeles, CA
- 3Medicine, Cardiology, Human Genetics, Microbiology, Immunology & Molecular Genetics, UCLA, Los Angeles, CA
- 4Anesthesiology, Physiology and Medicine, UCLA, Los Angeles, CA
Abstract 16236: Rescue of D2R Function in Mouse Kidney Using AAV9 Vector Abrogates the Renal Injury and High Blood Pressure Induced by D2R Silencing
Prasad Konkalmatt, Laureano D Asico, Yu Yang, Pedro A Jose, Ines Armando
Circulation. 2014;130:A16236
Abstract
The renal dopaminergic system plays a key role in salt homoeostasis and blood pressure regulation. Lack or down regulation of dopamine D2 receptor (D2R) function in mice increases the vulnerability to renal inflammation and high blood pressure. We hypothesized that rescuing renal D2R expression in mice in which D2R is silenced in the kidney will reduce renal inflammation, injury and normalize the blood pressure. Gene transfer studies showed that retrograde ureteral infusion of AAV9 vectors encoding firefly luciferase or eGFP provide heterologous gene expression in the collecting duct, distal convoluted tubule, and proximal tubule cells of the infused kidney. D2R expression was silenced by the renal sub-capsular infusion of D2R siRNA (D2RsiRNA) using osmotic mini pump (3 μg/day, n=6 per group). Mice treated with non-silencing siRNA (NS siRNA) served as controls. Fourteen days following the initiation of siRNA treatment, each group was treated with control AAV (CAAV) or AAV carrying wild-type D2R (D2RAAV) (n=3 per group, 1e+11 vgp/mouse). Fourteen days following AAV treatment, arterial blood pressure was measured and organs were collected. D2R expression was decreased in D2RsiRNA-treated kidneys compared with NS siRNA-treated kidneys (immunoblotting: 54 ± 0.8 vs 100± 22 %; P<0.05). D2RAAV treatment increased D2R expression (7.5-11-fold; P<0.01) in both D2RsiRNA and NS siRNA-treated mice. Mice treated with D2RsiRNA+CAAV had increased systolic blood pressure (121±3 mmHg; P<0.05) in comparison with the mice treated with D2RsiRNA+D2RAAV (100±6 mm Hg) (rescued mice), NS siRNA +CAAV (101±4 mm Hg), or NS siRNA +D2RAAV (101±1 mm Hg). D2R silencing (D2RsiRNA+CAAV) caused an increase in the expression of pro-inflammatory TNF-α, the pro-fibrotic TGF-β1 and its downstream target fibronectin-1, as well as kidney injury molecule-1 and ki-67, a marker of cell proliferation. By contrast, the expression of the same proteins was reversed to normal in the D2R-rescued group (D2RsiRNA+D2RAAV). In conclusion, these results demonstrate that increased blood pressure and renal injury due to D2R silencing could be rescued by over expression of D2R in the kidney using AAV9 vector. Furthermore, these data provide the basis for designing novel therapies for kidney disease.
Article Information
vol. 130 no. Suppl 2 A16236
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Prasad Konkalmatt;
- Laureano D Asico;
- Yu Yang;
- Pedro A Jose;
- Ines Armando
- Medicine, Univ of Maryland Sch of Medicine, Baltimore, MD
Abstract 15295: Molecular Mechanism of Chronic Sildenafil-Caused Regression of Heart Failure: Effects on the Express of Cardiac SR Ca2+-ATPase, Subtype of β-Adrenergic Receptors and Nitric Oxide Synthase Isoforms
Heng-Jie Cheng, Tiankai Li, Che Ping Cheng
Circulation. 2014;130:A15295
Abstract
Background: Sildenafil (SIL), a selective inhibitor of PDE5 has been shown to exert profound beneficial effects in heart failure (HF). Recently we further found that SIL caused regression of cardiac dysfunction in a rat model with isoproterenol (ISO)-induced progressive HF. However, the molecular basis is unclear. We hypothesized that reversal of HF-induced detrimental alterations on the expressions of cardiac SR Ca2+-ATPase (SERCA2a), β-adrenergic receptors (AR) and nitric oxide synthase (NOS) isoforms by SIL may play a key role for its salutary role in HF.
Methods: Left ventricular (LV) and myocyte function and the protein levels of myocyte β1- and β3- AR, SERCA2a, phospholamban (PLB) and three NOS were simultaneously evaluated in 3 groups of male rats (6/group): HF, 3 months (M) after receiving ISO (170 mg/kg sq for 2 days); HF/SIL, 2 M after receiving ISO, SIL (70 μg/kg/day sq via mini pump) was initiated and given for 1 M; and Controls (C).
Results: Compared with controls, ISO-treated rats progressed to severe HF at 3 M after ISO followed by significantly decreased LV contractility (EES, HF: 0.7 vs C: 1.2 mmHg/μl) and slowed LV relaxation, reductions in the peak velocity of myocyte shortening (77 vs 136 μm/sec), relengthening (62 vs 104 μm/sec) and [Ca2+]iT (0.15 vs 0.24) accompanied by a diminished myocyte inotropic response to β-AR agonist, ISO (10-8 M). These abnormalities were associated with concomitant significant decreases in myocyte protein levels of β1-AR (0.23 vs 0.64), SERCA2a (0.46 vs 0.80), PLBSer16/PLB ratio (0.24 vs 0.40) and eNOS (0.28 vs 0.46), but significantly increases in protein levels of β3-AR (0.29 vs 0.10) and iNOS (0.18 vs 0.08) with relatively unchanged nNOS. Chronic SIL prevented the HF-induced decreases in LV and myocyte contraction, relaxation, peak [Ca2+]iT, and restored normal myocyte contractile response to ISO stimulation. With SIL, protein levels of myocyte β1- and β3-AR, SERCA2a were restored close to control values, but eNOS was significantly elevated than controls (0.77).
Conclusions: Chronic SIL prevents HF-caused downregulation of cardiac β1-AR and reverse contrast changes between iNOS and β3-AR with SERCA 2a and eNOS expression, leading to the preservation of LV and myocyte function, [Ca2+]iT, and β-adrenergic reserve.
Article Information
vol. 130 no. Suppl 2 A15295
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Heng-Jie Cheng;
- Tiankai Li;
- Che Ping Cheng
- Internal Medicine-Cardiovascular Medicine, Wake Forest Sch of Medicine, Winston-Salem, NC
Abstract 13847: PTRF/Cavin-1 Knock-out Mice Develop a Progressive Cardiomyopathy with ERK1/2 Hyperactivation and Caveolin-3 Reduction
Takeru Kasahara, Takehiro Ogata, Naoki Maruyama, Takuya Taniguchi, Tetsuro Hamaoka, Kotaro Miyagawa, Naohiko Nakanishi, Tomomi Ueyama
Circulation. 2014;130:A13847
Abstract
Background: Mutations in the PTRF gene, coding for Cavin-1, cause congenital generalized lipodystrophy type 4 (CGL4) associated with myopathy. Symptoms of patients with CGL4 are more widespread comprising myopathy, smooth and skeletal muscle hypertrophy and osteopenia. In particular, patients with CGL4 due to PTRF/Cavin-1 mutations have fetal cardiac arrhythmia and long-QT syndrome. Patients with PTRF/Cavin-1 deficiency show reduction of Caveolin-3 (Cav3) whose deficiency leads to cardiac hypertrophy with loss of caveolae. However, it remains unknown whether loss of PTRF/Cavin-1 affects the phenotypic behavior cardiac myocytes in vitro. Here, we present a detailed characterization of the hearts of PTRF/Cavin-1 knock-out (KO) mice.
Methods and Results: PTRF/Cavin-1 KO mice developed a progressive cardiomyopathic phenotype. At four months of age, PTRF/Cavin-1 KO heats displayed significant wall thickness of left ventricles and reduced fractional shortening as revealed by echocardiography. Histological analysis revealed marked cardiomyocyte hypertrophy with progressive interstitial/peri-vascular fibrosis. Hypertrophy-related fetal gene expression was also induced in PTRF/Cavin-1 KO mice. Western blotting analysis and quantitative RT-PCR revealed that Cav3 expression was significantly suppressed in PTRF/Cavin-1 KO hearts compared with wild-type hearts. Because Cav3 deletion-induced cardiac hypertrophy shows hyperactivation of p42/44 MAPK (ERK1/2) and loss of caveolae in cardiomyocytes, we assessed the ERK1/2 activity and caveolar morphology in the heart of PTRF/Cavin-1 KO mice. ERK1/2 was hyperactivated in PTRF/Cavin-1 KO hearts. Furthermore, electron microscopy displayed the absence of caveolae in cardiomyocytes of PTRF/Cavin-1 KO mice. However, small vesicles were retained in PTRF/Cavin-1 KO cardiomyocytes.
Conclusion: PTRF/Cavin-1 KO mice develop a progressive cardiomyopathy with ERK1/2 hyperactivation. Our results suggest that this process is attributable to Cav3 reduction. Our data argue that loss of PTRF/Cavin-1 expression is sufficient to induce a molecular program leading to cardiomyocyte hypertrophy and cardiomyopathy.
Article Information
vol. 130 no. Suppl 2 A13847
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Takeru Kasahara;
- Takehiro Ogata;
- Naoki Maruyama;
- Takuya Taniguchi;
- Tetsuro Hamaoka;
- Kotaro Miyagawa;
- Naohiko Nakanishi;
- Tomomi Ueyama
- Cardiovascular Medicine, Graduate Sch of Med Science, Kyoto Prefectural Univer, Kyoto city, Japan
Abstract 12519: Regulation of Cardiac Fibroblast Phenotype by Scleraxis
Rushita Bagchi, Patricia Roche, Ronen Schweitzer, Michael P Czubryt
Circulation. 2014;130:A12519
Abstract
Cardiac fibroblasts constitute the primary extracellular matrix synthesis machinery in the myocardium. Activation of fibroblasts into a hyper-synthetic and contractile phenotype potentiates fibrosis, impairs cardiac function and contributes to heart failure. Our laboratory previously reported that the transcription factor scleraxis regulates human cardiac collagen Iα2 expression and has shown its up-regulation in the post-infarct scar. Here we demonstrate a novel regulatory role for scleraxis in governing cardiac fibroblast function and phenoconversion. Cell contractility assays using collagen gels demonstrated the abrogation of pro-fibrotic TGF-β-mediated contractility of myofibroblasts in response to scleraxis knockdown. The de novo expression of α-smooth muscle actin (αSMA) and its incorporation into stress fibers is a key feature of myofibroblasts – key causative cells of fibrosis. Scleraxis over-expression in isolated primary cardiac fibroblasts induced αSMA gene expression and stress fiber formation, and rescued the αSMA loss observed in cardiac fibroblasts from scleraxis null mice. Luciferase reporter assays demonstrated a significant transactivation of the αSMA gene promoter by scleraxis. Mutation analysis revealed that scleraxis interacts with two E-boxes within the αSMA promoter, a finding confirmed by chromatin immunoprecipitation of scleraxis in primary cardiac fibroblasts. An increase in scleraxis binding to the αSMA promoter was observed in cardiac myofibroblasts compared to fibroblasts, and also in response to TGF-β, further supporting a direct role of scleraxis in regulation of myofibroblast αSMA expression and its contractile phenotype. Gel shift assays also confirmed the direct interaction of scleraxis with E-boxes within the αSMA gene promoter. Our data indicates that scleraxis plays a required role in cardiac fibroblast phenotype and contractile function. Taken in context with our finding that scleraxis regulates expression of multiple extracellular matrix components, including fibrillar collagens, our data reveals that scleraxis exerts broad and potent pro-fibrotic effects on cardiac fibroblast form and function, and may thus represent a novel target for fibrosis therapy.
Article Information
vol. 130 no. Suppl 2 A12519
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Physiology & Pathophysiology, Institute of Cardiovascular Sciences/Univ of Manitoba, Winnipeg, Canada
- 2Cell and Developmental Biology, Oregon Health and Science Univ, Portland, OR
Abstract 11978: Post Infarct Treatment With Microrna145 Protects the Heart Against Myocardial Ischemia Reperfusion Injury Through Acceleration of Myocyte Autophagy
Kenshi Higashi, Yoshihisa Yamada, Minami Kumazaki, Takuma Aoyama, Shinya Baba, Shingo Minatoguchi, Kazuhiko Nishigaki, Genzou Takemura, Yukihiro Akao, Shinya Minatoguchi
Circulation. 2014;130:A11978
Abstract
Background: We previously reported that microRNA145 (miR-145) significantly reduced the myocardial infarct size and improved the function of left ventricle.
It has been reported that autophagy is activated in cardiomyocytes in ischemic heart disease. We investigated whether miR-145 would reduce the myocardial infarct size through acceleration of autophagy.
Objective: We aimed to investigate whether administration of miR-145 would affect myocyte autophagy in a rabbit model of myocardial infarction (MI).
Methods: Male Japanese white rabbits underwent 30 min of coronary occlusion followed by 2 and 14 days of reperfusion. Saline or miR-145 (0.035 mg/kg) was intravenous injected immediately after reperfusion. Some of the rabbits treated with miR-145 were administered chloroquine that acts by inhibiting vacuolar H+-ATPase and autophagosome fusion to prevent the final digestion step during autophagy immediately after reperfusion followed by 30 min of coronary ligation. On day 2 and day 14 post-MI, rabbits were sacrificed and the hearts were removed. Heart tissues were sampled and devided into three area; remote area, borderline area and infarct area. The morphological changes were assessed by electron microscopy, and the expressions of LC3B, Akt, phosphorylated (p)-Akt, ERK and p-ERK were assessed by western blot analysis.
Results: The MI size as a percentage of area at risk was significantly smaller in the miR-145 group than that in the control group. Treatment with chloroquine blocked the infarct size-reducing effect of miR-145. These were observed both in 2days and 14days reperfusion groups. Electron microscopic findings showed the autophagic change in cardiomyocytes in both the control and miRNA145 groups on day 2 and day 14 post-MI. Western blot analysis showed that transition from LC3B-I to LC3B-II was significantly stronger in the miR-145 group than in the control group. The expression of p-Akt in the ischemic area was upregulated in the miR-145 group on day 2 post-MI.
However, there was no significant difference in the expression of Akt, ERK and p-ERK between 2 groups on day 2 and day 14 post-MI.
Conclusions: It is suggested that post-infarct treatment with miR-145 attenuated ischemia-reperfusion injury through acceleration of myocyte autophagy.
Article Information
vol. 130 no. Suppl 2 A11978
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Kenshi Higashi1;
- Yoshihisa Yamada1;
- Minami Kumazaki2;
- Takuma Aoyama1;
- Shinya Baba1;
- Shingo Minatoguchi1;
- Kazuhiko Nishigaki1;
- Genzou Takemura1;
- Yukihiro Akao3;
- Shinya Minatoguchi1
- 1Faculty of medicine, Gifu Univ Graduate Sch Of Medicine, Gifu, Japan
- 2United Graduate Sch of Drug Discovery and Med information Science, Gifu university, Gifu, Japan
- 3United Graduate Sch of Drug Discovery and Med information Science, Gifu Univ, Gifu, Japan
Abstract 12008: Nos1 Adapter Protein Nos1ap Overexpression Alters Qtc Intervals in Transgenic Mice
Tatjana Williams, Anahi P Arias-Loza, Marco Abeßer, Joachim Schmitt, Kai Schuh, Oliver Ritter
Circulation. 2014;130:A12008
Abstract
Background: Congenital long- or short-QT syndrome may lead to life-threatening ventricular tachycardia and sudden cardiac death. Apart from rare disease-causing mutations in ion channels, common genetic variations in the neuronal nitric oxide synthase (NOS1) regulator NOS1AP, have recently been associated with QT interval variations in a human whole-genome association study. In fact, NOS1AP SNPs have been linked to increases in QTc intervals and sudden cardiac death. We therefore speculate that myocardial NOS1AP overexpression may lead to a decrease of the QTc interval and an increased susceptibility to rhythm disorders.
Methods and Results: We generated transgenic mice (TG) with a conditional myocardial NOS1AP overexpression and focused on electrical alterations. Conditional overexpression of NOS1AP resulted in a 147% ventricular increase in TG mice compared to WT littermates. NOS1AP was mainly located at the sarcolemma where it interacted with NOS1 and the L-type Ca2+- channel. HW/BW ratio, ventricular ANP expression, ventricular cross-sectional area and collagen deposition were not altered in NOS1AP mice under baseline conditions.
However, NOS1AP overexpressing mice showed a clear decrease of QTc intervals (33 vs. 48 ms). They were more prone to bradycardia (resting heart rate 467 bpm vs. 666 bpm). Atrial programmed stimulation repeatedly caused atrial tachycardia. Ventricular programmed stimulation caused VT in some mice with NOS1AP overexpression.
We also investigated the functional effect of the human rs16847548 (T/C). We found that this SNP decreased NOS1AP promoter activity in a viral NOS1AP luciferase assay, suggesting that this SNP downregulates NOS1AP expression in humans.
Conclusion: Myocardial overexpression of NOS1AP leads to a significant shortening of the QTc interval with an increased susceptibility to atrial and ventricular rhythm disorders. SNP rs16847548 in NOS1AP resulted in downregulation of NOS1AP expression which provides an explanation for elongation of QTc intervals. In summary, not only a mutation in ion channels itself but also genetic alterations in expression of ion channel modifiers, such as NOS1AP, have an impact on QTc intervals.
Article Information
vol. 130 no. Suppl 2 A12008
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Dept of Medicine I, Univ Hosp of Wuerzburg, Wuerzburg, Germany
- 2Dept of Physiology, Univ of Wuerzburg, Wuerzburg, Germany
- 3Dept of Medicine I, Univ of Wuerzburg, Wuerzburg, Germany
- 4Comprehensive Heart Failure Cntr, Univ Hosp of Wuerzburg, Wuerzburg, Germany
Abstract 20344: CCN5 Reverses Fibrosis and Cardiac Dysfunction Induced by Pressure Overload in Murine Models
Dongtak Jeong, Min-Ah Lee, Changwon Kho, Ahyoung Lee, Jae Gyun Oh, Jason Kovacic, Woo Jin Park, Roger Hajjar
Circulation. 2014;130:A20344
Abstract
Introduction: Cardiac fibrosis (CF) is associated with increased stiffness and diastolic dysfunction in failing hearts, and its severity is an independent predictor for clinical outcomes of heart failure patients. We previously showed that a matricellular protein CCN5 is anti-fibrotic and anti-hypertrophic in the mouse heart. However, the mechanisms involved in the anti-fibrotic activity of CCN5 remains unknown.
Methods: In this study, we tested two different animal models. First, we generated pressure-overload heart failure models in mouse by TAC operation. Two months following gene transfer of CCN5, cardiac function was evaluated by echocardiography and invasive hemodynamics. Second, endothelial cells were recently shown to significantly contribute to CF through Endothelial-Mesenchymal Transition (EndoMT). Thus, we tested whether CCN5 inhibits EndoMT using the Scl-Cre-ERT; R26RstopYFP double Tg mouse, which is a widely used lineage traceable animal model. Protein and RNA expression levels of CCN5, several types of collagen and onventional TGF-beta signaling related genes were measured by western blot and RT-PCR analysis. Cell growth assay and apoptosis assay were also performed to evaluate the function of CCN5 in isolated rat adult cardiomyocytes and non-cardiomyocyte cells. Third, EndoMT and transdifferentiation assays were performed using human coronary endothelial cells (HCEC) and myofibroflast in vitro.
Results: First, CCN5 induces accelerated degradation of preformed fibrogenic materials like collagen in the heart. CCN5 reduces the fraction of Vimentin-positive fibroblasts that have already expanded in response to TAC. Second, our data showed that CCN5 can inhibit EndoMT both in vivo and in vitro. Finally, we found that CCN5 selectively induced apoptosis in myofibroblasts but not in cardiomyocytes or fibroblasts.
Conclusions: CCN5 evoked reversal of preformed CF, which was accompanied by functional recovery of the failing hearts through the inhibition of transdifferentiation into myofibroblast and selective induction of apoptosis on myofibroblasts. Taken together, our results that CCN5 can effectively target fibrosis in the setting of heart failure.
Article Information
vol. 130 no. Suppl 2 A20344
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Dongtak Jeong1;
- Min-Ah Lee2;
- Changwon Kho1;
- Ahyoung Lee1;
- Jae Gyun Oh1;
- Jason Kovacic1;
- Woo Jin Park3;
- Roger Hajjar1
- 1Cardiology, Mount Sinai Sch of Medicine, New York, NY
- 2life Science, Gwangju Institute of Science and Technology, Gwangju, Korea, Republic of
- 3Life Science, Gwangju Institute of Science and Technology, Gwangju, Korea, Republic of
Abstract 18865: Identification of a Functional SNP Regulating PRRX1 at the 1q24 Locus for Atrial Fibrillation
Elena Dolmatova, Nathan R Tucker, Honghuang Lin, Rebecca R Cooper, Jiangchuan Ye, Moritz F Sinner, Maxim Imakaev, Steven A Lubitz, Jordan Leyton-Mange, Gus Vlahakes, Emelia J Benjamin, Kathryn L Lunetta, Leonid Mirny, David J Milan, Patrick T Ellinor
Circulation. 2014;130:A18865
Abstract
Introduction: Genome-wide association studies have identified 9 genomic loci associated with atrial fibrillation (AF).
Hypothesis: We sought to identify the functional variant at the 1q24 locus for AF, located upstream of the paired related homeobox 1 gene (PRRX1).
Methods: We used morpholino-mediated knockdown in zebrafish to assess the role of PRRX1 in cardiac function and development. To identify potential enhancers at the PRRX1locus we analyzed DNase hypersensitivity, histone methylation, and mammalian conservation data from ENCODE. Tissue-specific enhancer activity was evaluated by microinjection of eGFP reporter constructs for each putative enhancer into zebrafish and luciferase assays in a mouse atrial myocyte (HL-1) cell line. To determine physical interaction between the AF-associated enhancer and PRRX1 promoter we analyzed available Hi-C data and performed chromatin conformation capture (3C). The functional SNP was localized using luciferase assays in HL-1 cells. The effect of the functional SNP on gene expression in human left atrial tissue was measured by qPCR.
Results: Knockdown of the PRRX1 ortholog in zebrafish resulted in atrial dilation and shortening of atrial action potential duration (APD80: 114.8±2.2ms vs 126±1.5ms in controls, p=0.0004). Of the 4 regions tested at the 1q24 locus, 2 adjacent regions exhibited enhancer activity in the zebrafish myocardium. 3C demonstrated an increased interaction frequency between the enhancer and PRRX1 promoter regions in cells of cardiac lineage when compared to controls (103±57%, p=0.038). Screening for functional SNPs within these regions revealed that the AF risk allele (C) at SNP rs577676 associated with ~4 fold increased enhancer activity as compared to the non-risk (T) allele in HL-1 cells. Finally, regional eQTL analysis of human atrial tissue showed that rs577676 correlated with PRRX1expression.
Conclusions: We have implicated PRRX1 in cardiac electrophysiology by demonstrating that knockdown of the gene results in atrial dilation and shortening of atrial action potential duration. Further, we have found that SNP rs577676 modifies an enhancer regulating PRRX1 expression.
Article Information
vol. 130 no. Suppl 2 A18865
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Elena Dolmatova1;
- Nathan R Tucker1;
- Honghuang Lin2;
- Rebecca R Cooper1;
- Jiangchuan Ye1;
- Moritz F Sinner3;
- Maxim Imakaev4;
- Steven A Lubitz1;
- Jordan Leyton-Mange1;
- Gus Vlahakes5;
- Emelia J Benjamin6;
- Kathryn L Lunetta7;
- Leonid Mirny4;
- David J Milan1;
- Patrick T Ellinor1
- 1CVRC, MGH, Charlestown, MA
- 2Boston Univ Sch of Medicine, Computational Biomedicine Section, Dept of Medicine, Boston, MA
- 3CVRC, Ludwig-Maximilians-Univ, Campus Grosshadern,, Munich, Germany
- 4Institute for Med Engineering and Sciences,, Massachusetts Institute of Technology, Cambridge, MA
- 5Dept of Surgery, MGH, Boston, MA
- 6Preventive Medicine Section, Dept of Medicine, Boston Univ Sch of Medicine, Boston, MA
- 7Dept of Biostatistics,, Boston Univ Sch of Public Health, Boston, MA
Abstract 13867: A Genome-wide Association Study of Nonsyndromic Mitral Valve Prolapse and Functional Studies of Risk Loci Provide Insight Into Underlying Biological Mechanisms
Nabila Bouatia-Naji, Christian DINA, Nathan TUCKER, Francesca N DELLING, Katelynn TOOMER, Ronen DURST, Maelle PERROCHEAU, Leticia FERNANDEZ-FRIERA, Jorge SOLIS, Thierry LE TOURNEAU, Ming-Huei CHEN, Vincent PROBST, Yohan BOSSE, Philippe PIBAROT, Mark LATHROP, Serge HERCBERG, Ronan ROUSSEL, Fabrice BONNET, Richard REDON, Hervé LE MAREC, Philippe FROGUEL, Ramachandran S VASAN, Patrick BRUNEVAL, Russell A NORRIS, David J MILAN, Susan A SLAUGENHAUPT, Robert A LEVINE, Jean-Jacques SCHOTT, Albert A HAGEGE, Xavier JEUNEMAITRE
Circulation. 2014;130:A13867
Abstract
Background: Nonsyndromic mitral valve prolapse (MVP) is a common, progressively degenerative valvulopathy. MVP is the most frequent indication for surgical repair of mitral regurgitation, and causes heart failure, arrhythmia and sudden death. However, the genetic basis and physiopathology of MVP remain elusive. The genetic heterogeneity and high prevalence (2-5%) of MVP support the utility of a genome-wide association study in large populations to identify risk loci, which may uncover novel pathogenic mechanisms.
Methods: We tested 4.8 million common genotyped/imputed variants in 1412 sporadic MVP cases and 2439 controls. Replication for 23 loci was performed in 4 case control studies from USA, France, Spain and Canada, all of European ancestry (Ncases=1442, NControls=6779). High profile candidate genes were investigated for protein expression by immunohistochemistry on mitral valves in embryonic and adult mice and morpholino knockdown (KD) assessed cardiac function in zebrafish.
Results: Six susceptibility loci were identified after replication (P<5х10-8). Association with MVP was observed in LMCD1 (OR=1.32, P =1.3х10-11), a repressor of GATA6 previously implicated in cardiac hypertrophy. Morpholino knockdown of Lmcd1 in zebrafish resulted in a significant atrioventricular (AV) valve defect with regurgitation. Another signal on Chr2q35 (OR=1.25, P=3.1х 10-11) mapped upstream to TNS1, which encodes a focal adhesion and actin interacting protein. We found that tensin1 is expressed during valve morphogenesis in mice and maintained in the adult endothelial and valvular interstitial cells. Interestingly, Tns1-/- mice exhibited enlarged posterior mitral leaflets compared to wild type. In addition, zebrafish knockdown of Tns1 (and not Igfbp5 or Igfbp2 in the same human locus) induced AV regurgitation.
Conclusions: In this multidisciplinary study we discovered 6 loci with effect sizes (OR range: 1.22-1.33) compatible with a complex genetic pattern of inheritance, and identified new actors in valve development and biology. We provide genetic and functional evidence implicating LMCD1 and TNS1 in valve development and function. This study reveals new pathways that can potentially be modified to improve the natural history of MVP.
Article Information
vol. 130 no. Suppl 2 A13867
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Nabila Bouatia-Naji1;
- Christian DINA2;
- Nathan TUCKER3;
- Francesca N DELLING4;
- Katelynn TOOMER5;
- Ronen DURST6;
- Maelle PERROCHEAU7;
- Leticia FERNANDEZ-FRIERA8;
- Jorge SOLIS8,
- PROMESA investigators;
- Thierry LE TOURNEAU9;
- Ming-Huei CHEN10;
- Vincent PROBST9;
- Yohan BOSSE11;
- Philippe PIBAROT12;
- Mark LATHROP13;
- Serge HERCBERG14;
- Ronan ROUSSEL15;
- Fabrice BONNET16;
- Richard REDON2;
- Hervé LE MAREC17;
- Philippe FROGUEL18;
- Ramachandran S VASAN19;
- Patrick BRUNEVAL20;
- Russell A NORRIS5;
- David J MILAN21;
- Susan A SLAUGENHAUPT22;
- Robert A LEVINE23,
- the Leducq Transatlantic MITRAL Network;
- Jean-Jacques SCHOTT2;
- Albert A HAGEGE24,
- MVP-France Investigators;
- Xavier JEUNEMAITRE25
- 1Paris Descartes Univ, PRES Sorbonne Paris Cité, INSERM UMR970 Paris Cardiovascular Rsch Cntr, Hôpital Européen Georges Pompidou, Paris, France
- 2Université de Nantes, Inserm UMR1087, CNRS UMR 6291, Institut du Thorax, Cntr Hospier Universitaire (CHU) Nantes,, Nantes, France
- 3Cntr for Human Genetic Rsch, Massachusetts General Hosp, Cardiovascular Rsch Cntr, Massachusetts General Hosp, Charlestown, MA
- 4Dept of Medicine (Cardiovascular Div), Beth Israel Deaconess Med Cntr, Harvard Med Sch, Boston, MA
- 5Dept of Regenerative Medicine and Cell Biology, Cardiovascular Developmental Biology Cntr, Children’s Rsch Institute, Med Univ of South Carolina, Charleston, SC
- 6Cardiology Dept, Hadassah Hebrew Univ Med Cntr, Jerusalem, Israel
- 7Paris Descartes Univ, PRES Sorbonne Paris Cité, NSERM UMR970 Paris Cardiovascular Rsch Cntr, Hôpital Européen Georges Pompidou, Paris, France
- 8Hosp Universitario Monteprincipe, Cntr Nacional de Investigaciones Cardiovasculares (CNIC), Madrid, Spain
- 9Cntr Hospier Universitaire (CHU), Inserm UMR1087, CNRS UMR 6291, Institut du Thorax, Cntr Hospier Universitaire (CHU) Nantes,, Nantes, France
- 10Dept of Neurology, Boston Univ Sch of Medicine, Framingham Heart Study, Boston, MA
- 11Institut universitaire de cardiologie et de pneumologie de Québec, Laval Univ, Laval, Canada
- 12Université de Laval, Québec Heart & Lung Institute, Laval, Canada
- 13Cntr d’étude du polymorphisme humain, CNG Fondation Jean Dausset, Paris, France
- 14Nutritional Epidemiology Rsch Team, Epidemiology and biostatistics Cntr, Dept of Public, Paris 13 Univ, Sorbonne Paris Cité, Cnam, Paris 5 Univ, Paris 7 Univ, Bobigny, France
- 15Endocrinologie, Diabétologie et Nutrition, AP-HP, Hôpital Bichat, INSERM, UMRS 1138, Cntr de Recherche des Cordeliers,, Paris, France
- 16Dept of Endocrinology, Univ Hosp Rennes, Inserm UMR 991, Université Rennes 1, Rennes, France
- 17Cntr Hospier Universitaire (CHU) Nantes, INserm UMR1087, CNRS UMR 6291, Institut du Thorax, Nantes, France
- 18Lille 2 Univ, European Genomic Institute for Diabetes, CNRS UMR 8199, Lille Pasteur Institute, Lille, France
- 19Framingham Heart Study, Framingham Heart Study, Framingham, MA
- 20APHP-Dept of Pathology, INSERM UMR970 Paris Cardiovascular Rsch Cntr, Paris, France
- 21Cardiology Dept, Cardiovascular Rsch Cntr, Massachusetts General Hosp, Charlestown, MA
- 22Cntr for Human Genetic Rsch, Massachusetts General Hosp, Harvard Med Sch, Boston, MA
- 23Cardiac Ultrasound Laboratory, Massachusetts General Hosp and Harvard Med Sch, Boston, MA
- 24Dept of Cardiology, Paris Descartes Univ, PRES Sorbonne Paris Cité, AP-HP Hôpital Européen Georges Pompidou, Paris, France
- 25AP-HP Dept of Genetics, Pars Descartes Univ, INSERM UMR970 Paris Cardiovascular Rsch Cntr, Hôpital Européen Georges Pompidou, Paris, France
Abstract 12389: Epigenome-Wide Study Identifies Novel Methylation Loci Associated With Body Mass Index and Waist Circumference
Stella Aslibekyan, Ellen W Demerath, Michael Mendelson, Degui Zhi, Weihua Guan, Liming Liang, Jin Sha, James S Pankow, Chunyu Liu, M. Ryan Irvin, Myriam Fornage, Bertha Hidalgo, Li-An Lin, Krista C Stanton Thibeault, Jan Bressler, Michael Y Tsai, Megan L Grove, Paul N Hopkins, Eric Boerwinkle, Ingrid B Borecki, Jose M Ordovas, Daniel Levy, Hemant K Tiwari, Devin M Absher, Donna K Arnett
Circulation. 2014;130:A12389
Abstract
Introduction: Body mass index (BMI) and waist circumference (WC) are complex heritable traits with direct implications for human health. An emerging body of evidence links epigenetic processes such as DNA methylation to gene regulation and obesity phenotypes.
Hypothesis: We hypothesized that methylation patterns across the genome are associated with variation in adiposity traits, specifically BMI and WC.
Methods: To identify specific genomic regions that play a role in the epigenetics of obesity, we quantified epigenome-wide DNA methylation in CD4+ T-cells using the Illumina Infinium HumanMethylation450 array in 991 European American participants of the Genetics of Lipid Lowering Drugs and Diet Network. We subsequently conducted association analyses, modeling percent methylation at individual cytosine-phosphate-guanine (CpG) sites as a function of BMI and WC, adjusting for age, gender, study site, T-cell purity, smoking, and family structure.
Results: We found epigenome-wide significant associations between eight CpG sites and BMI and five CpG sites and WC, and successfully replicated the top hits in whole blood samples from 2,377 European American participants of the Framingham Heart Study and 2,105 African American participants of the Atherosclerosis Risk in Communities study. Top findings were located in CPT1A (meta-analysis P= 3.5×10-37 for BMI and P=2.2×10-16 for WC), PHGDH (meta-analysis P= 4.7×10-15 for BMI and 2.2×10-8 for WC), CD38 (meta-analysis P= 3.7×10-11 for BMI and 6.1×10-13 for WC) and long intergenic non-coding RNA 00263 (meta-analysis P= 1.2×10-13 for BMI and 5.8×10-10 for WC), regions with biologically plausible relationships to adiposity.
Conclusions: This large-scale epigenome-wide study identified novel robust associations between DNA methylation at CpG loci and obesity indices, laying the groundwork for future diagnostic and/or therapeutic applications.
Article Information
vol. 130 no. Suppl 2 A12389
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Stella Aslibekyan1;
- Ellen W Demerath2;
- Michael Mendelson3;
- Degui Zhi4;
- Weihua Guan5;
- Liming Liang6;
- Jin Sha1;
- James S Pankow2;
- Chunyu Liu7;
- M. Ryan Irvin1;
- Myriam Fornage8;
- Bertha Hidalgo1;
- Li-An Lin9;
- Krista C Stanton Thibeault10;
- Jan Bressler9;
- Michael Y Tsai11;
- Megan L Grove9;
- Paul N Hopkins12;
- Eric Boerwinkle8;
- Ingrid B Borecki13;
- Jose M Ordovas14;
- Daniel Levy3;
- Hemant K Tiwari4;
- Devin M Absher10;
- Donna K Arnett1
- 1Epidemiology, Univ of Alabama at Birmingham, Birmingham, AL
- 2Div of Epidemiology and Community Health, Univ of Minnesota, Minneapolis, MN
- 3Population Sciences Branch, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD
- 4Biostatistics, Univ of Alabama at Birmingham, Birmingham, AL
- 5Div of Biostatistics, Univ of Minnesota, Minneapolis, MN
- 6Epidemiology, Harvard Sch of Public Health, Boston, MA
- 7Biostatistics, Boston Univ, Boston, MA
- 8Brown Foundation Institute of Molecular Medicine, The Univ of Texas Health Science Cntr, Houston, TX
- 9Human Genetics Cntr, The Univ of Texas Health Science Cntr, Houston, TX
- 10-, Hudson Alpha Institute for Biotechnology, Huntsville, AL
- 11Div of Laboratory Medicine and Pathology, Univ of Minnesota, Minneapolis, MN
- 12Internal Medicine, Univ of Utah, Salt Lake City, UT
- 13Div of Statistical Genomics, Univ of Washington in St. Louis, St Louis, MO
- 14Jean Mayer USDA HNRCA, Tufts Univ, Boston, MA
Abstract 20207: Deciphering and Exploiting MicroRNA-Target Interactomes in Human Cardiac Tissues
Congsheng Cheng, Ryan M Spengler, Frances L Johnson, Stravros G Drakos, Dean Y Li, Nikos Diakos, Beverly L Davidson, Ryan L Boudreau
Circulation. 2014;130:A20207
Abstract
The orchestration of complex biological functions requires gene regulatory networks that are modulated, in part, by microRNAs (miRNAs). During the past decade, miRNAs have emerged as key biological effectors, and growing evidence indicates that miRNAs play critical roles in an array of human diseases, including cardiovascular conditions. These noncoding RNAs associate with Argonaute proteins (e.g. Ago2) to direct post-transcriptional gene suppression via base-pairing with target transcripts. To better understand how miRNAs contribute to protective and pathogenic responses to disease, identifying their targets in affected tissues is of paramount importance. Here, we addressed the latter by profiling Ago2:RNA interactions using crosslinking immunoprecipitation coupled with high-throughput sequencing (CLIP-seq) to generate the first transcriptome-wide map of miRNA binding sites in human heart tissues (normal and failing, n=4 each). We uncovered thousands of Ago2 binding sites which are highly enriched for conserved sequences corresponding to abundant cardiac miRNAs. This interactome provides a valuable resource for accelerating our understanding of miRNA functions in the cardiovascular system. Notably, our initial exploration of these data has revealed numerous clinically-relevant interactions involving miRNAs and target genes previously implicated in cardiomyopathies (e.g. miR-1, -23, -24, -29, -208, Serca2, Ryr2, CaMKII, among others). Also, the interactome points to a coordination of miRNA activities in controlling calcium handling and signaling, mitochondrial function, and metabolic signaling pathways. Overall, this work represents an initial step towards characterizing the diverse landscape of miRNA:target interactions across normal and diseased human cardiac tissues, and the primary data offers clues which may facilitate the translation of genetic studies of complex cardiovascular-related diseases into novel or refined pathogenic mechanisms and therapeutics.
Article Information
vol. 130 no. Suppl 2 A20207
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Congsheng Cheng1;
- Ryan M Spengler1;
- Frances L Johnson1;
- Stravros G Drakos2;
- Dean Y Li2;
- Nikos Diakos2;
- Beverly L Davidson1;
- Ryan L Boudreau1
- 1Internal Medicine, Univ of Iowa, Iowa City, IA
- 2Internal Medicine, Univ of Utah, Salt Lake City, UT
Abstract 20419: Citrullination in Actin-Tropomyosin-Myosin Complex: Novel Regulatory Mechanism
Justyna Fert-Bober, Jennifer Van Eyk
Circulation. 2014;130:A20419
Abstract
Background: A novel protein post-translational modification, citrullination was shown previously in a number of key myofilament proteins, tropomyosin (R 133, R 238), actin (R 39) and myosin heavy chain (R 1176, 1303, 1434) in HF patient (values for total spectra counts for citrullinated proteins in control, ISHD and IDCM: 1.8 ±1.3, 3.2±2.7 and 2.3±1.9, respectively). The alterations in contractile proteins underlying enhanced Ca2+-sensitivity of the contractile apparatus in end-stage failing human myocardium are still not resolved. Protein citrullination arises from the enzymatic conversion of arginine residues to citrulline result in loss of a positive charge and reduction in hydrogen-bonding ability. And here, the biophysical and biochemical effect on myofilament function is determined.
Method: F-actin-tropomyosin binding, tropomyosin-actin-myosin, actin-myosin and myosin ATPase activity assays, and F-actin stability assays were carried out.
Results: In vitro citrullinated tropomyosin significantly enhance affinity for F-actin (p=0.001) and decrease in the ATPase activity (p=0.06). Furthermore, citrullination of myosin HMM is not essential for actin affinity, although it modulate ATPase activation (p=0.3). Contrary, the ATPase activity is increase by pretreatment of actin (but not myosin) with PAD2 (p=0.09).
Conclusion: Citrullination of the contractile proteins could affect different aspects of regulatory function. It either triggers a structural change or stabilizes a conformation that is necessary for actin-activated release of Pi and completion of the ATPase cycle.
Significance: Citrullination of specific myofilament proteins in HF can have dramatic effect on modulating actin filament integrity and myosin function and tropomyosin action on myofilament regulation.
Article Information
vol. 130 no. Suppl 2 A20419
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Heart institute, Cedars-Sinai Med Cntr, Los Angeles, CA 90048, CA
- 2Heart institute, Cedars-Sinai Med Cntr, Los Angeles, CA
Abstract 18382: Rna Splicing Regulated by A2bp1 is Essential for Cardiac Function in Zebrafish
Karen S Frese, Benjamin Meder, Andreas Keller, Jan Haas, Steffen Just, Hugo A Katus, Wolfgang Rottbauer
Circulation. 2014;130:A18382
Abstract
Objective: Alternative splicing (AS) is one of the key mechanisms for the proteomic and functional diversity of eukaryotes. However, the complex nature of AS, its associated regulators and their targets are only partially understood. In the present study we investigated the transcriptomic diversity in the zebrafish heart using RNA-Sequencing and elucidated the functional role of the splicing regulator A2BP1 in vivo.
Results: Using RNA-Sequencing we characterized the cardiac transcriptome of 48 hours post fertilization (hpf) old zebrafish embryos and compared the expression of genes and their isoforms to whole fish tissue. Besides the known cardiac genes, we found several previously described genes, highly expressed in cardiac tissue. The analysis of RNA-Seq data indicates that 14% of all genes expressed in the heart undergo AS by single exon-skipping/inclusion. To determine the effect of splicing factors on mRNA splicing we investigated the functional role of splicing regulator a2bp1 in vivo by using the zebrafish as a model organism. Morpholino-mediated a2bp1 knockdown in zebrafish embryos led to progressive cardiac contractile dysfunction, suggesting an important role of a2bp1 in maintenance of cardiac function. Splicing analysis revealed that loss of a2bp1 does not result in a completely splicing failure, but rather alters the splicing pattern of specific target genes. Here we identified novel spliceforms and potentialy novel targets of splicing factor a2bp1. Splice-junction blockage experiments showed that a balanced isoform expression of the targets actn3a, hug, ktn1, ptpla and camk2g is necessary for maintaining cardiac function in zebrafish. We assume, that the a2bp1-knockdown phenotype is not caused by missplicing of specific targets rather by the cumulative effect of many splicing abnormalities.
Conclusion: Our study reveal a novel splicing regulator that is necessary for normal heart function. We showed that dysfunction of a2bp1 not only leads to heart failure, but show that a2bp1 mediates the splicing of different transcripts which might mediate the observed phenotype. Our results highlight the importance of balanced mRNA splicing in the heart and represents intriguing opportunities for novel therapeutic approaches.
Article Information
vol. 130 no. Suppl 2 A18382
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Karen S Frese1;
- Benjamin Meder1;
- Andreas Keller2;
- Jan Haas1;
- Steffen Just3;
- Hugo A Katus1;
- Wolfgang Rottbauer3
- 1Med III, Univ Hosp Heidelberg, Heidelberg, Germany
- 2Bioinformatics, Saarland Univ, Saarbrücken, Germany
- 3Dept of Internal Medicine II, Univ Hosp Ulm, Ulm, Germany
Abstract 17782: Both Common and Rare SCN10A Variants Associated with Brugada Syndrome Displayed an Increase in Late Nav1.8 Sodium Currents in ND 7/23 cells
Eleonora Savio-Galimberti, Tao Yang, Dan M Roden, Elijah R Behr, Kaylen C Kor, Yalda Jamshidi, Evmorfia Petropoulou, Pascale Guicheney, Jacob Tfelt-Hansen, Arthur A Wilde, Connie R Bezzina, Morten S Olesen, Anders G Holst, Michael J Ackerman, Peter J Schwartz, Lia Crotti, Vincent Probst, Richard Redon, Dawood Darbar
Circulation. 2014;130:A17782
Abstract
Introduction: Brugada syndrome (BrS) is an oligogenic disease, often linked to mutations in SCN5A encoding the canonical cardiac sodium channel. Prolongation of QRS interval implicates slowed cardiac conduction as an important element of the BrS arrhythmia phenotype. Recent genome-wide association studies have implicated common variation in SCN10A as a potential modulator of cardiac conduction. SCN10A encodes the tetrodotoxin-resistant voltage-gated sodium channel isoform Nav1.8 primarily found in dorsal root ganglia and at lower levels in the heart. A recent candidate gene sequencing study identified 5 non-synonymous SCN10A rare variants in 4/156 white SCN5A mutation-negative patients with BrS and one protective non-synonymous common variant V1073A [(T>C): T allele BrS vs. control: 65.1% vs. 40.1%, P = 3.54×10-19].
Hypothesis: Here we tested the hypothesis that the common variant (V1073A) and 2 of the rare variants identified (A200V, I671V) generate aberrant Nav1.8 function and this may contribute to BrS phenotype.
Methods: We studied the SCN10A common variant V1073A and the SCN10A rare variants A200V and I671V in transiently transfected ND7/23 cells. After 48-hr incubation at 37°C, macroscopic sodium currents were measured at room temperature using whole-cell voltage-clamp technique.
Results: V1073A common variant demonstrated two-fold increase in both peak (INa-Peak) and late (INa-L, measured 100-ms post-depolarization) sodium currents compared to WT. By contrast, both rare variants demonstrated significant reductions in INa-Peak (A200V: -16.5±3 pA/pF [mutant] vs. -37.8±4.9 pA/pF [WT], P<0.01; I671V: -25.5±1.6 pA/pF [mutant] vs. -37.8±4.9 pA/pF [WT], P<0.05, all n=7 cells/group) but increased INa-L (A200V: 24.2±3.3% of INa-Peak; I671V: 15.8±1.6% of INa-Peak) compared to WT (7.8±1.3% of INa-Peak). Incubation of cells transfected with either A200V or I671V rare variants at 28oC instead of 37oC did not rescue the reduction observed in peak currents.
Conclusions: SCN10A variants associated with BrS displayed a range of strikingly altered functions in a heterologous expression system (ND7/23 cells), which support our hypothesis that they contribute to BrS phenotype.
Article Information
vol. 130 no. Suppl 2 A17782
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Eleonora Savio-Galimberti1;
- Tao Yang1;
- Dan M Roden1;
- Elijah R Behr2;
- Kaylen C Kor1;
- Yalda Jamshidi2;
- Evmorfia Petropoulou2;
- Pascale Guicheney3;
- Jacob Tfelt-Hansen4;
- Arthur A Wilde5;
- Connie R Bezzina5;
- Morten S Olesen6;
- Anders G Holst6;
- Michael J Ackerman7;
- Peter J Schwartz8;
- Lia Crotti8;
- Vincent Probst9;
- Richard Redon9;
- Dawood Darbar1,
- Brugada Gene Discovery Group
- 1Medicine, Vanderbilt Univ, Nashville, TN
- 2Cardiology, St. George’s Univ of London, London, United Kingdom
- 3Institut de recherche sur les malaries cardiovasculaires, du metabolisme et de la nutrition, Faculte de Medecine Pierre et Marie Curie, Paris, France
- 4Cardiology, Copenhagen Univ Hosp, Copenhagen, Denmark
- 5Clinical and Experimental Cardiology, Univ of Amsterdam, Amsterdam, Netherlands
- 6Laboratory of Molecular Cardiology, The Heart Cntr, Copenhagen Univ Hosp, Copenhagen, Denmark
- 7Cardiology, Mayo Clinic, Rochester, MN
- 8Cntr for Cardiac Arrhythmias of Genetic Origin, IRCCS Istituto Auxologico Italiano, Milano, Italy
- 9L’institut du Thorax, Universite de Nantes, Nantes, France
Abstract 17545: Different Human Mutations in the Myosin Binding Protein C3 (MYBPC3) Produce Specific Cardiac Phenotypes in the Zebrafish
Sahar I Da’as, Joseph Yu, Jonathan T Butcher, Navaneethakrishnan Krishnamoorthy, Jassim Al Suwaidi Al Suwaidi, Heba Kassem, Kholoud N Al Shafai, Mohammed A Al-Hashemi, Lama Shuayb, Thomas Brand, Magdi H Yacoub
Circulation. 2014;130:A17545
Abstract
Mutations in the gene encoding myosin binding protein C3 (MYBPC3) are one of the commonest causes of Hypertrophic cardiomyopathy and can produce varying phenotypes. The exact disease mechanisms responsible remain unknown. Zebrafish model offers unique opportunities to study human cardiovascular disease mechanisms in vivo.
We have modeled different human MYBPC3 mutations in zebrafish embryos. Morpholinos targeting zebrafish mybpc3 were injected to model four disease causing missense mutations of domain C1. Mutation1 (Arg177His), Mutation 2 (Ala216Thr), Mutation 3 (Glu258Lys) that were identified in Egypt for the first time by Kassem et al [1] and Mutation 4 (Ser217Gly) that was recently identified in Qatar. The morpholino targets Mutation 1, 2 and 4 at exon 5 and Mutation 3 at exon 6 splice donor site in zebrafish embryos, in order to precisely recapitulate the human mutations.
Different MYBPC3 human mutations produced specific cardiac phenotypes in zebrafish embryos. These morphant embryos (3 days old) displayed aberrant cardiac phenotype and induced hypertrophic cardiomyopathy similar to the human phenotype. Defective heart phenotype was observed in 51% of Mutation 1, 2 and 4 and 68% of Mutation 3. In both groups pericardial edema was present in almost 20%; as an early manifest of heart failure. Mutation 3 resulted in severe cardiac phenotype exhibited by more zebrafish morphant embryos with enlarged cardiac chambers and reduced heart rate compared to other mutations. These results support our molecular modeling of domain C1, suggesting that Mutations 1, 2 and 4 increase intra-molecular rigidity to induce structural changes and have minimal effect on electrostatic properties at the surface. Interestingly, Mutation 3 inversely impacts the structural properties and has major effect on the surface of C1 and may lead to malfunction of the protein.
In summary: we investigated features that characterize human MYBPC3 variants in zebrafish model. The translational site blocking of zebrafish mybpc3 recapitulated the human hypertrophic cardiomyopathy, while missense Mutation 3 at exon 6 seems to cause more severe disease phenotype than Mutation 1, 2 and 4 at exon 5 in the zebrafish morphant embryos.
- Kassem H. et al 2013 J Cardiovasc Transl Res 6: 65-80
Article Information
vol. 130 no. Suppl 2 A17545
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Sahar I Da’as1;
- Joseph Yu2;
- Jonathan T Butcher3;
- Navaneethakrishnan Krishnamoorthy1;
- Jassim Al Suwaidi Al Suwaidi1;
- Heba Kassem4;
- Kholoud N Al Shafai1;
- Mohammed A Al-Hashemi5;
- Lama Shuayb1;
- Thomas Brand2;
- Magdi H Yacoub1
- 1Qatar Cardiovascular Rsch Cntr, Qatar Foundation, Doha, Qatar
- 2Heart Science Cntr, Imperial College London, London, United Kingdom
- 3Dept of Biomedical Engineering, Cornell Univ, Ithaca, NY
- 4Alexandria Faculty of Medicine, Aswan Heart Cntr, Alexandria, Egypt
- 5Hamad Med Corp, Heart Hosp, Doha, Qatar
Abstract 17200: Altered Nuclear and Cytoskeletal Mechanics and Defective Cell Adhesion in Cardiac Myocytes Carrying the Cardiomyopathy LMNA D192G Mutation
Orfeo Sbaizero, Thomas Lanzicher, Valentina Martinelli, Carlin Long, Dobrmir Slavov, Giorgia Del Favero, Matthew Taylor, Luisa Mestroni
Circulation. 2014;130:A17200
Abstract
Introduction: Previous investigations suggested that lamin A/C gene (LMNA) mutations, which cause a variety of human diseases including muscular dystrophies and dilated cardiomyopathy, alter the nuclear mechanical properties.
Hypothesis: We hypothesized that not only the nucleus, but also the whole-cell biomechanical behavior may be altered in cardiomyocytes with the dilated cardiomyopathy LMNA D192G mutation.
Methods: We combined atomic force microscopy (AFM), molecular and cellular biology methodologies to study the biomechanics of the nucleus and whole-cell. Neonatal rat ventricular myocytes (NRVMs) were infected with adenoviral vectors containing either wild type or LMNA D192G. LMNA protein expression was confirmed up to day 6. Nuclear and whole-cell biomechanics were investigated in LMNA D192G, wild type and control NRVMs, .
Results: Live-cell AFM force-deformation curves from days 1 through 6 showed that LMNA D192G nuclei displayed higher stiffness and fragility compared to controls with a peak at 72h (P<0.05), with 3 time increase in nuclear Young modulus. Furthermore, mutant NRVMs showed a severe reduction in the adhesion area between AFM probe and cell membrane compared to control and wild type. Finally, D192G NRVMs displayed increased viscoelasticity behavior measured as force decays with time during cell deformation (relaxation force test) compared to wild type and control NRVMs, suggesting loss of cytoskeleton elasticity. The altered biomechanical behavior of LMNA D192G NRVMs was rescued by wild-type LMNA (P<0.05).
Conclusions: Our study suggests that the cardiomyopathy LMNA D192G mutation has a profound effect on the whole-cell biomechanics in cardiomyocytes, extending beyond the increased nuclear stiffness and fragility, and involving cytoskeletal structural modifications and reduced cell membrane adhesion, changes that can be rescued by wild-type LMNA.
Article Information
vol. 130 no. Suppl 2 A17200
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Orfeo Sbaizero1;
- Thomas Lanzicher1;
- Valentina Martinelli2;
- Carlin Long3;
- Dobrmir Slavov3;
- Giorgia Del Favero1;
- Matthew Taylor3;
- Luisa Mestroni3
- 1Engineering, Univ of Trieste, Trieste, Italy
- 2Molecular Medicine, I.C.G.E.B., Trieste, Italy
- 3Cardiovascular Institute, Univ of Colorado Denver AMC, Aurora, CO
Abstract 16314: Identification of MicroRNAs Inducing Adult Cardiomyocyte Proliferation
Raghav Pandey, Laeia Jackson, Gang Ma, Rafeeq p Ahmed
Circulation. 2014;130:A16314
Abstract
Background: Even though neonatal cardiomyocytes (NCM) proliferate robustly, adult cardiomyocytes (ACM) have very little proliferative potential. By functional screening of 875 microRNAs (miRs) Eulalio et al 2012 identified 46 miRs which induce proliferation of NCMs by more than 35%. Additional studies by Mahmood et al 2013 have identified Meis1 as the major player that controls ACM cell cycle. We hypothesize that the proliferation inducing potential of miRs may vary between NCMs and ACMs and some of these miRs may induce proliferation through Meis1 regulation.
Methods and Results: ACM were isolated from male rats aged 4-6 months and transfected with miR mimics for cel-67 (control) and 24 of the top NCM proliferation inducing miRs individually. Edu was administered for 4 days starting day 1 after transfection. Cells were fixed on day 5 and immuno-stained. Proliferating cardiomyocytes were identified by co-staining of TnI and Edu and the percentage was calculated in each group. Eighteen of the 24 miRs induced significant proliferation of ACMs . The most significant of them with more than 7% increase in ACM proliferation were miR-302b-5p (7.8±0.6), miR-1910 (9.8±0.76), miR-548c-3p (11.9±2.7), miR-2053 (10.1±0.4), miR-936 (11.6±3.4), miR-1825 (54±5.2), miR-509 (7.1±0.62), miR-590 (11.5±1.1) and miR-23b-3p (12.7±0.35). Of these we identified 3 miRs (miR-548c-3p, miR-509-3p and miR-23b) to have binding site on the 3’UTR of Meis1 and simultaneously showing an increase in proliferation. Meis1 has been established as a critical transcriptional regulator of CM proliferation through activation of CDK inhibitors.
Conclusion: To the best of our knowledge this is the first study to identify a panel of miRs inducing ACM proliferation. Delivering these miRs to the infarcted region is a promising approach and has the potential to regenerate the ischemic heart by inducing proliferation of CMs surrounding the infarct zone.
Article Information
vol. 130 no. Suppl 2 A16314
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Pathology, Univ of Cincinnati, Cincinnati, OH
- 2Pathology, Meharry Med College, Nashville, TN
Abstract 15411: Estrogen Receptor β Mediates Sex Differences in the Proteomic Response of the Heart to Pressure Overload
Georgios Kararigas, Daniela Fliegner, Stefanie Forler, Oliver Klein, Carola Schubert, Joachim Klose, Vera Regitz-Zagrosek
Circulation. 2014;130:A15411
Abstract
Sex differences in the response of the heart to pressure overload (PO) have pointed to estrogen receptor (ER) β, which may be cardioprotective. However, the underlying mechanisms are incompletely defined. In a proteomic analysis of PO-induced hypertrophy, we hypothesized significant sex differences mediated by ERβ. Two-month-old C57BL6 male (M) and female (F) wild-type (WT) and ERβ knockout (BERKO) mice were subjected to transverse aortic constriction (TAC) or sham surgery (n = 4/group). The proteome of left ventricular samples was separated by high-resolution 2-dimensional polyacrylamide gel electrophoresis followed by mass spectrometry. The proteomic data were analyzed with empirical Bayesian-based linear models using R/Bioconductor. Nine weeks after TAC, heart-weight-to-tibia-length ratio was significantly higher in WT M (TAC vs. sham 128%; P < 0.05) than in WT F (TAC vs. sham 54%; P < 0.05) mice (interaction P < 0.001), while this difference in BERKO mice between M (TAC vs. sham 53%; P < 0.05) and F (TAC vs. sham 55%; P < 0.05) was abolished (interaction P = 0.5). Following quality assessment, background correction and normalization of the proteomic data, 795 left ventricular protein spots were identified and analyzed. Our comparative proteomic analysis revealed that in WT M and F mice 82 and 31 protein spots, respectively, differed between sham and TAC (P ≤ 0.05). In BERKO M and F mice we found 114 and 87 altered protein spots, respectively (P ≤ 0.05). Mitochondrial bioenergetics-related proteins, such as trifunctional enzyme subunit family members, were repressed in M-WT-TAC mice, while cytoskeletal proteins were induced in F-WT-TAC mice. On the other hand, heat shock proteins were induced in both M and F-BERKO-TAC mice. Candidates in metabolic, mitochondrial and cytoskeletal pathways were selected for validation of the proteomic analysis by means of Western blotting. We conclude that ERβ mediates sex-specific remodeling playing a major role in the proteomic response of the heart to PO. In particular, metabolic and mitochondrial proteins are repressed in males but maintained in females. We expect that our study will be a useful resource for the unraveling of the effects of sex and ERβ in PO.
Article Information
vol. 130 no. Suppl 2 A15411
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Georgios Kararigas1;
- Daniela Fliegner1;
- Stefanie Forler2;
- Oliver Klein3;
- Carola Schubert1;
- Joachim Klose3;
- Vera Regitz-Zagrosek1
- 1Institute of Gender in Medicine & Cntr for Cardiovascular Rsch, Charite Univ Hosp, Berlin, Germany
- 2Institute for Human Genetics, Charite Univ Hosp, Berlin, Germany
- 3Core Unit Proteomics, Berlin-Brandenburg Cntr for Regenerative Therapies, Charite Univ Hosp, Berlin, Germany
Abstract 15454: Inhibition of Mechanistic Target of Rapamycin by Heterozygous Deletion of Raptor Ameliorates Pressure-overload Induced Heart Failure and the Associated Proteomics Remodeling
Dao Fu Dai
Circulation. 2014;130:A15454
Abstract
Background: We reported that inhibition of mechanistic target of rapamycin (mTOR) by short-term rapamycin or caloric restriction ameliorates age-dependent cardiac hypertrophy and diastolic dysfunction. Although inhibition of mTOR signaling is well known to regulate metabolism and suppress protein synthesis, the mechanisms of beneficial effect of mTOR inhibition in cardiac hypertrophy and failure are not fully understood.
Method: To investigate the mechanisms underlying beneficial effect of mTOR inhibition, we used the transverse aortic constriction (TAC)-induced heart failure model and examined the effect of heterozygous deletion of Raptor (Raptor het), a component of mTOR complex 1, and transgenic overexpression of cardiac specific wild type or mutant 4EBP1, one of the main downstream target of mTOR complex 1. Global proteomics analysis was performed using an improved label-free quantitative shotgun approach, followed by Ingenuity Pathway analysis.
Results: In wild-type mice with TAC-induced heart failure, global proteomics analysis revealed decreased abundance of proteins involved in mitochondrial function, electron transport chain, citric acid cycle and fatty acid metabolism and increased abundance of proteins involved in several signaling pathways (RhoA, actin, integrin) as well as oxidative stress response and protein ubiquitin pathways. Raptor het attenuate TAC induced heart failure, accompanied by better preservation of proteomics remodeling, especially the proteins involved in mitochondrial function, citric acid cycle and protein ubiquitination pathways. In contrast, either transgenic overexpression of wild type 4EBP1 or mutant 4EBP1 abolish the adaptive hypertrophy in response to TAC by suppressing protein translation, and thereby aggravate heart failure, in parallel with adverse remodeling of left ventricular proteomes. Neonatal cardiomyocyte experiments reveal that PGC1-α and Sirt3 are among the candidate signaling mechanisms linking the mTOR inhibition and mitochondrial metabolism.
Conclusion: mTOR inhibition by Raptor heterozygous deletion, but not overexpression of 4EBP1, ameliorates TAC-induced heart failure and associated with better preservation of mitochondrial proteome.
Article Information
vol. 130 no. Suppl 2 A15454
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Dao Fu Dai
- Pathology, Univ of Washington, Seattle, WA
Abstract 20140: Minibrain Relate Kinase / Dyrk1B Links Skeletal Muscle Glycolytic Metabolism with Insulin Resistance and Causes Metabolic Syndrome
Mohsen Fathzadeh, Ali Reza Keramati, Gwang Go, Rajvir Singh, Kazem Sarajzadeh, Javad Tavakkoly-Bazzaz, Ali Noorafshan, Mohammad Kasaei, Masoud Amini, Gholam Hossein R Omrani, Mohammad Ali Babaee Bigi, Masoud Babaei, Adalat Hosseinian, Reza Malekzadeh, Richard Lifton, Arya Mani
Circulation. 2014;130:A20140
Abstract
We have identified a novel nonconservative mutation in Minibrain related serine/threonine kinase (Mirk/ Dyrk1B) in outlier kindreds with metabolic syndrome. The mutation substitutes cysteine for arginine (R102C) and segregates with most traits of metabolic syndrome, including central obesity, diabetes and hypertension. Oral glucose tolerance test (OGTT) in young nondiabetic mutation carriers revealed insulin resistance compared to noncarrier family members. Since skeletal muscle (SM) is the largest organ for glucose uptake and metabolism, we obtained Vastus Lateralis biopsies of mutation carriers and their unaffected relatives and examined them for gene/protein expression by deep RNA sequencing (RNA-Seq) and Western blot analysis and for fiber composition by immunostaining. The fiber composition data demonstrated fewer slow-twitch fibers (35% vs. 75%) and more fast -twitch fibers (65% vs. 25%) in SM of mutation carriers vs. controls. Interestingly, there were increased protein expression levels of fast-twitch fiber type proteins (MYH11, MYLPF), pyruvate dehydrogenase kinase, pyruvate kinase, and neuronal nitric oxide synthase in SM of mutation carriers vs. noncarriers. Consistent with these findings, the protein expression levels of the master regulator of cellular energy metabolism mitochondrial biogenesis, PPAR-gamma coactivator (PGC-1a), were reduced and the nuclear expression levels of FOXO1 and NFAT were increased. Similar findings were observed when wildtype and mutant (R102C) Dyrk1B were overexpressed in C2C12 cells. The overexpression of the kinase deficient Dyrk1B (Y271/273F) similarly resulted in reduced expression of PGC-1a and increased expression of nuclear FOXO1, suggesting kinase independent effects. Taken together, these findings suggest that enhanced kinase-independent activities of Dyrk1B, either through increased expression or by its gain of function mutation R102C induce insulin resistance by promoting glycolytic metabolism and reducing oxidative phosphorylation. In conclusion, Dyrk1B is a potential target for development of novel drugs that aim to enhance skeletal muscle insulin sensitivity.
Article Information
vol. 130 no. Suppl 2 A20140
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Mohsen Fathzadeh1;
- Ali Reza Keramati1;
- Gwang Go1;
- Rajvir Singh1;
- Kazem Sarajzadeh2;
- Javad Tavakkoly-Bazzaz3;
- Ali Noorafshan2;
- Mohammad Kasaei2;
- Masoud Amini2;
- Gholam Hossein R Omrani2;
- Mohammad Ali Babaee Bigi2;
- Masoud Babaei4;
- Adalat Hosseinian4;
- Reza Malekzadeh5;
- Richard Lifton1;
- Arya Mani1
- 1Internal Medicine, Yale Univ, New Haven, CT
- 2Medicine, Shiraz Univ of Med Sciences, Shiraz, Iran, Islamic Republic of
- 3Med Genetics, Theran Univ of Med Sciences, Tehran, Iran, Islamic Republic of
- 4Medicine, Ardabil Univ of Med Sciences, Ardabil, Iran, Islamic Republic of
- 5Director Digestive Disease Rsch Institute, Theran Univ of Med Sciences, Tehran, Iran, Islamic Republic of
Abstract 18864: A Genetic Variant of Human Aldosterone Synthase Causes Salt-Dependent High Blood Pressure in Transgenic Mice
Brahmaraju Mopidevi, Nitin Puri, Meenakshi Kaw, Sudhir Jain, Narsimha Keetha, Steven Fiering, Ashok Kumar
Circulation. 2014;130:A18864
Abstract
Aldosterone, synthesized by the enzyme CYP11B2, induces positive sodium-balance and predisposes to hypertension. Various investigators, using genomic DNA analyses, have linked -344T polymorphism in the hCYP11B2 gene to human hypertension. We have identified three SNPs, in linkage disequilibrium, in the hCYP11Be gene: T/A at -663, T/C at -470 and C/T at -344. Variants ACT occur together and form the haplotype I (Hap I) while variants TTC constitute haplotype II (Hap II). We hypothesize that these SNPs, when present together, will lead to haplotype-dependent transcription of the hCYP11B2 gene, differentially increase aldosterone and affect blood pressure. To this end, novel transgenic (TG) mice with the hCYP11B2 gene, targeted to the mHPRT locus, with either haplotype II or I variant are used in the study. ChIP assay, using anti-RNA pol II antibody, shows increased Pol II binding to the chromatin from Hap I TG mice in adrenal (2.8 fold higher, p<0.05) and renal tissues (1.3 fold higher, p<0.05) as compared to chromatin extracts from Hap II-TG mice. Immunoblot analysis shows upregulation of the hCYP11B2 in adrenal (2.7 fold higher, p<0.05) and renal tissues (1.35 fold higher, p<0.05) of Hap I vs. Hap II-TG mice; no significant difference was observed in mCYP11B2 between the two haplotypes. Complementary ELISA shows higher circulating levels (p<0.05) of aldosterone in Hap I mice (1504±48.7 pg/mL) as compared to both, Hap II (778±142.8 pg/mL) and C57 mice (740±28.9 pg/mL). Importantly, we observed increased baseline blood pressure in Hap I TG mice (Hap I- 117±2.5 vs. Hap II- 109±1.9 mm Hg, p<0.05), an effect accentuated by high-salt diet (Hap I- 135±2.6 vs. Hap II- 122±2.2 mm Hg, p<0.05). Elevated aldosterone was accompanied by up-regulation (p<0.05) of proinflammatory markers in renal tissues from Hap I-TG mice (IL1β, MCP1, ICAM). Thus, this study identifies -344T as a reporter polymorphism for Hap I of the hCYP11B2 gene. SNPs in Hap I promote increased transcription and expression of the gene, in multiple tissues, with resultant elevation of plasma aldosterone levels. Pathophysiological impact of this haplotype-dependent transcriptional regulation of the hCYP11B2 is highlighted by increased inflammation and blood pressure in TG mice with the Hap I of this transgene.
Article Information
vol. 130 no. Suppl 2 A18864
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Brahmaraju Mopidevi1;
- Nitin Puri1;
- Meenakshi Kaw1;
- Sudhir Jain1;
- Narsimha Keetha1;
- Steven Fiering2;
- Ashok Kumar1
- 1Physiology and Pharmacology, The Univ of Toledo, Toledo, OH
- 2Microbiology and Immunology, Dartmouth Med Sch, Lebanon, NH
Abstract 18010: Induced Pluripotent Stem Cell-Based Modeling of Human NOTCH1 Mutations Reveals Novel Pathways Regulating Aortic Valve Disease
Christina V Theodoris, Mark P White, Molong Li, Lei Liu, Daniel He, Katherine S Pollard, Benoit G Bruneau, Deepak Srivastava
Circulation. 2014;130:A18010
Abstract
In humans, NOTCH1 mutations result in congenital heart defects including valve malformations and severe valve calcification in adults. Valve calcification typically occurs on the aortic side of the valve leaflets that is not exposed to laminar shear stress, suggesting a protective role of flow sensed by the endothelial lining. To understand the mechanisms by which NOTCH1 mutations in endothelial cells (ECs) cause disease, we generated induced pluripotent stem cell (iPSC) lines from fibroblasts of four individuals from two families affected with aortic valve disease due to heterozygous non-sense mutations in NOTCH1. We differentiated control and mutant iPSC lines into ECs and exposed the ECs to either static or fluid shear stress conditions. NOTCH1 mRNA levels were decreased by ~50% in the NOTCH1+/- ECs under static or shear stress conditions. RNA-seq revealed that 165 genes were differentially expressed in static conditions and 193 genes responded abnormally to shear stress in NOTCH1+/- ECs compared to NOTCH1+/+ ECs. Differentially expressed genes included canonical NOTCH1 targets HRT2 and EFNB2 as well as novel targets involved in vascular development, inflammation, and endochondral ossification. Anti-calcific genes uniquely upregulated in shear stress were dysregulated in NOTCH1+/- ECs, indicating that they were unable to mount the normal protective response induced by shear stress in the valve. Generating isogenic NOTCH1+/+ and NOTCH1+/- cell lines using TALEN genome editing identified genes specifically dysregulated due to NOTCH1 heterozygosity rather than differentially expressed due to genetic background and showed rescue of this dysregulation in TALEN-corrected NOTCH1+/+ ECs. We have mapped the gene networks dysregulated in NOTCH1+/- ECs as determined by NOTCH1 ChIP-seq, differentially methylated regions of DNA, and genome-wide differences in the progression of activating and repressive chromatin states that begin to explain the mechanisms by which heterozygosity of a transcription factor leads to disease-specific changes. Determining the consequence of NOTCH1 heterozygous mutations in human patient-specific ECs will reveal novel mechanisms underlying aortic valve disease, leading to potential targets for intervention.
Article Information
vol. 130 no. Suppl 2 A18010
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Christina V Theodoris1;
- Mark P White1;
- Molong Li1;
- Lei Liu1;
- Daniel He1;
- Katherine S Pollard1;
- Benoit G Bruneau2;
- Deepak Srivastava1
- 1Gladstone Institute of Cardiovascular Disease, Gladstone Institutes, Univ of California, San Francisco, San Francisco, CA
- 2Gladstone Institute of Cardiovascular Disease, Gladstone Insitutes, Univ of California, San Francisco, San Francisco, CA
Abstract 16391: Constitutively Active GSK-3β Attenuates Cardiac Aging Through Ulk1-Dependent Stimulation of Autophagy
Peiyong Zhai, Yoshiyuki Ikeda, Akihiro Shirakabe, Takanobu Yamamoto, Bonaventure Magrys, Xin Cheng, Junichi Sadoshima
Circulation. 2014;130:A16391
Abstract
Phosphorylation of glycogen synthase kinase-3β (GSK-3β) at S9 increases during aging or cellular senescence. However, it is unknown whether the phosphorylation is beneficial or detrimental, and mechanisms through which GSK-3β affects aging remain to be elucidated. Autophagy, which protects organs against aging, decreases with aging. We hypothesize that GSK-3β mediates its effects via Ulk1, a regulator of autophagy, and studied unphosphorylatable/constitutively active (CA) GSK-3βS9A knock-in mice (βKI) and GSK-3βS9A/Ulk1+/- bigenic mice (Bigenic) up to 24 months (M) of age. From 6M to 24M, left ventricular (LV) weight/body weight (LVW/BW, mg/g) increased in wild-type mice (WT, 3.2±0.2 vs 3.8±0.2) but decreased in βKI (3.2±0.3 vs 2.4±0.1), and was lower (p<0.005) in βKI than in WT at 24M. Cardiac myocyte cross-sectional area (CSA, μm2) was smaller in βKI than in WT at 24M (376±5 vs 501±8, p<0.001). The LVW/BW was higher (3.5±0.2, p<0.001) and the CSA was bigger (527±4, p<0.001) in Bigenic than in βKI at 24M. These data suggest that CA GSK-3β attenuates aging-related cardiac hypertrophy via Ulk1. Cardiac fibrosis (%) increased at 24M in WT (3.5±0.2 vs 1.1±0.1, p<0.001), but was less in βKI (2.4±0.1) than in WT (p<0.001) or Bigenic (5.5±0.6, p<0.001). The TUNEL-positive nuclei (%) increased at 24M in WT (0.17±0.02 vs 0.03±0.01, p<0.001), but there were fewer in βKI (0.04±0.01) than in WT (p<0.005) or Bigenic (0.19±0.03, p<0.005). These data demonstrate that CA GSK-3β decreases aging-associated cardiac fibrosis and apoptosis via Ulk1. The LV end-systolic elastance (mmHg/μl) decreased in WT (5±1 vs 16 ±2, p<0.005) but not in βKI, while the LV chamber stiffness constant (μl-1) increased in WT (0.13±0.0 vs 0.04±0.01, p<0.001) but not in βKI, and these effects of CA GSK-3β were abolished in Bigenic. These data suggest that CA GSK-3β preserves cardiac function via Ulk1. The numbers of autophagosomes and autolysosomes, determined using tandem fluorescent mRFP-GFP-LC3 transgenic mice (tfLC3), were significantly higher in βKI/tfLC3 mice than in tfLC3 or βKI/tfLC3/Ulk1+/- mice, indicating that CA GSK-3β activates autophagy via Ulk1. In conclusion, S9 phosphorylation of GSK-3β is detrimental during aging, and CA GSK-3β beneficially enhances Ulk1-dependent autophagy.
Article Information
vol. 130 no. Suppl 2 A16391
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Peiyong Zhai;
- Yoshiyuki Ikeda;
- Akihiro Shirakabe;
- Takanobu Yamamoto;
- Bonaventure Magrys;
- Xin Cheng;
- Junichi Sadoshima
- Cell Biology and Molecular Medicine, Rutgers-New Jersey Med Sch, Newark, NJ
Abstract 16184: Single Cell RNA Sequencing Reveals Dynamic and Heterogeneous Changes of Transcriptome During Cardiac Differentiation in vitro
shunsuke funakoshi, Masaki Nomura, Chikako Okubo, Kenji Miki, Takeshi Kimura, Shinya Yamanaka, Akira Watanabe, Yoshinori Yoshida
Circulation. 2014;130:A16184
Abstract
Human induced pluripotent stem cells-derived cardiomyocytes (iPSC-CMs) are expected to be clinically applicable, especially in the field of disease modeling and drug screening. However, one of the major problems to be overcome is their immaturity and heterogeneity, because these problems hinder the acquisition of stable experimental results for the clinical use. To overcome these problems, we performed the single cell RNA sequencing during cardiac differentiation in vitro and explored deeply the insights underlining cardiac differentiation. We successfully defined the differentiation state of cells at single cell level by principle component analysis (PCA) and largely recapitulate the cardiac development process with dynamic changes of transcriptome during the differentiation. In addition, by combination with weighted gene co-expression network analysis (WGCNA), we found out that known core transcriptional factors, such as TBX5, GATA4, MEF2C, and Nkx2.5, were co-regulated in the primitive CMs, but were differently regulated in the late differentiation stage, implying that a novel mechanism regulated the cardiac maturation processes. In addition, PCA during cardiac differentiation clarified the heterogeneous development of iPSC-CMs and we identified a novel surface marker, by which we could enrich well-matured CMs from heterogeneous population of differentiated CMs. Taken together, these results suggested that high resolution analyses by single cell RNA sequencing could be applicable not only to deeply understand the cardiac differentiation processes, but also to discover a novel important gene regulation for understanding the immaturity and heterogeneity during differentiation in vitro.
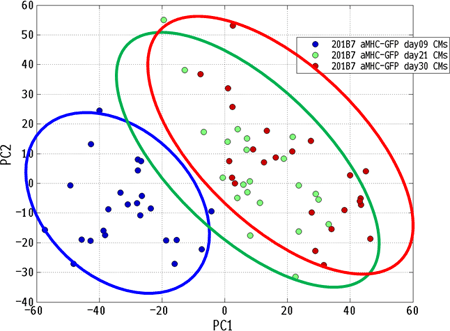
Article Information
vol. 130 no. Suppl 2 A16184
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- shunsuke funakoshi1;
- Masaki Nomura2;
- Chikako Okubo2;
- Kenji Miki2;
- Takeshi Kimura1;
- Shinya Yamanaka2;
- Akira Watanabe2;
- Yoshinori Yoshida2
- 1Cardiovasular medicine, Kyoto Univ, Kyoto, Japan
- 2Reprogramming Science, Cntr for iPS Cell Rsch and Application,Kyoto university, Kyoto, Japan
Abstract 20500: Trx1 Promotes Trans-Nitrosylation of Cellular Proteins to Stimulate Autophagy and Cell Survival
Narayani Nagarajan, Sebastiano Sciarretta, Junichi Sadoshima
Circulation. 2014;130:A20500
Abstract
Thioredoxin-1 (Trx1) is cardioprotective during oxidative stress, mainly through its antioxidant activity. Trx1 is also S-nitrosylated and, in turn, trans-nitrosylates other proteins. However, the role of Trx1-dependent S-nitrosylation in cardiomyocytes (CMs) is unknown. Here, we investigated the role of Trx1-mediated protein S-nitrosylation in the regulation of CM survival during stress in vitro. Using biotin-switch assays, we found that wild-type Trx1 (Trx1WT) is S-nitrosylated at baseline, but the extent of S-nitrosylation was attenuated in Trx1C73S, suggesting that Trx1 is S-nitrosylated at Cys73. Trx1WT and Trx1C73S do not differ in their redox activity, as determined by Amplex Red assays. Cellular protein S-nitrosylation levels were increased after 4 hours of glucose deprivation (GD), an energy stress condition (1.64±0.27 fold, p<0.05), as determined by biotin switch assays. Overexpression of Trx1WT increased (3.94-fold), whereas knockdown of Trx1 (0.66±0.01 fold, p<0.01) or overexpression of Trx1C73S (0.77±0.02 fold, p<0.01) decreased, total protein S-nitrosylation in response to GD. These results suggest that Trx1C73 regulates protein S-nitrosylation in CMs during GD. Overexpression of Trx1 increased CM survival after 24 hours of GD (1.42±0.08 fold vs LacZ, p<0.05), as evaluated with propidium iodide assays. Conversely, shTrx1 (2.13±0.05 fold vs control, p<0.01) or Trx1C73S (1.73±0.034 fold vs LacZ, p<0.01) increased cell death during GD. Either knockdown of Trx1 (LC3-II/Tubulin: 0.55 fold vs control) or overexpression of Trx1C73S (vs LacZ: LC3-II/Tubulin, 0.60 fold; autophagosomes, 0.83±0.16-fold, p<0.005; autolysosomes, 0.62±0.13-fold, p<0.005) significantly decreased autophagy during GD. Mechanistically, Trx1 co-immunoprecipitates with Atg7, an E1-like protein which plays a critical role in mediating autophagy. Using mass spectroscopy analyses, we found that SNO-Trx1 can trans-nitrosylate Atg7 in vitro. These results suggest that Trx1 trans-nitrosylates Atg7 during GD. Taken all together, our results indicate that Trx1 promotes trans-nitrosylation of cellular proteins, including Atg7, and autophagy, thereby promoting cell survival during energy stress in CMs.
Article Information
vol. 130 no. Suppl 2 A20500
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Narayani Nagarajan;
- Sebastiano Sciarretta;
- Junichi Sadoshima
- Cell Biology and Molecular Medicine, Rutgers Sch of Biomedical Sciences, Rutgers Univ, Newark, NJ
Abstract 18239: Yy1 Expression is Sufficient for the Mantenance of Cardiac Progenitor Cell State
Guang Li, Serge Gregoire, Anthony C. Sturzu, Robert J. Schwartz, Sean M. Wu
Circulation. 2014;130:A18239
Abstract
BACKGROUND: Transcription factors and epigenetic modifiers regulate gene expression during cardiac development. We have previously shown that YY1 is essential for the commitment of mesodermal precursors into cardiac progenitor cells (CPCs); however, the role of YY1 in the maintenance of CPC phenotype and their differentiation into cardiomyocytes is unknown.
METHODS AND RESULTS: We performed genome-wide expression analysis and targeted real-time qPCR as well as chromatin immunoprecipitation assays for histone modification in mouse embryonic stem cell (ESC)-derived CPCs with and without YY1 over-expression. We found, by genome-wide transcriptional profiling and phenotypic assays, that YY1 overexpression prevents cardiomyogenic differentiation and maintains the proliferative capacity of CPCs. We show further that the ability of YY1 to regulate CPC phenotype is associated with its ability to modulate histone modifications specifically at a developmentally critical enhancer of Nkx2-5 and other key cardiac transcription factors such as Tbx5. Specifically, YY1 overexpression promotes the maintenance of markers of gene activation, such as the acetylation of histone H3 at lysine 9 (H3K9Ac) and lysine 27 (H3K27Ac), as well as trimethylation at lysine 4 (H3K4Me3) at the Nkx2-5 cardiac enhancer. Furthermore, transcription factors associated proteins such as PoII, p300, and Brg1 are also enriched at the Nkx2-5 enhancer with YY1 overexpression. The biological activities of YY1 in CPCs appear to be cell-autonomous, based on co-culture assays in differentiating CPCs that over-express YY1.
CONCLUSION: These results demonstrate that YY1 expression is sufficient to maintain a CPC phenotype through its ability to sustain the presence of activating epigenetic and chromatin marks at key cardiac enhancers.
Article Information
vol. 130 no. Suppl 2 A18239
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Stanford, Cardiovascular Institute, Stanford, CA
- 2Dept of Medicine, Cardiovascular Rsch Cntr, Massachusetts General Hospita, CA
- 3Univ of Houston, Texas Heart Institute and Cntr for Molecular Medicine and Experimental Therapeutics, Houston, TX
Abstract 16797: Mechanisms That Repress BMP2 in Mesenchymal Cells
Melissa Rogers, Anastasios Fotinos, Tapan Shah
Circulation. 2014;130:A16797
Abstract
BMP2 is an essential protein. A short burst of BMP2 promotes repair after vascular injury. However, excessive BMP2 levels in the vasculature and heart valves promote pathological calcification. A sequence within the 3’UTR of the mRNA termed the “ultra-conserved sequence” (UCS) has been largely unchanged since fishes and mammals diverged. Cre-lox mediated deletion of the UCS in a mouse reporter transgene revealed that one role of the UCS is to repress BMP2 synthesis in heart valves and vascular cells. We are now testing the hypothesis that normal heart morphology requires UCS-mediated restraint of BMP2 synthesis. We used homologous recombination to generate a Bmp2 allele with the endogenous Bmp2 UCS flanked by loxP sites. We bred this strain to strains that express Cre-recombinase thus deleting the UCS. The survival of pups with homozygous loss of the UCS was significantly reduced. We are now assessing embryonic heart morphology in these strains. We also postulate that the regulatory proteins and microRNAs that mediate UCS-mediated repression may be exploited to pharmacologically reawaken BMP2 repression in tissues undergoing pathological pathologies. Specific miRNAs inhibit UCS-bearing reporters and mutations of miRNA binding sites activate these reporters. Interestingly, an A+U rich element (ARE) known to bind HuR (generally an activator) and AUF1 (a repressor) is embedded in a conserved miRNA binding site. Selective mutations of the ARE and the miRNA seed indicate that repressive molecules bind this site in mesenchymal cells where the UCS functions as a repressor.
Article Information
vol. 130 no. Suppl 2 A16797
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Melissa Rogers;
- Anastasios Fotinos;
- Tapan Shah
- Biochemistry & Molecular Biology, Rutgers NJ Med Sch, Newark, NJ
Abstract 16977: MicroRNA-mediated Regulation of SUMO1 Expression in Heart
Jae Gyun Oh, Ahyoung Lee, Dongtak Jeong, Alessia Baccarini, Brian D Brown, Changwon Kho, Roger J Hajjar
Circulation. 2014;130:A16977
Abstract
Background: Cardiac sarcoplasmic reticulum calcium ATPase pump (SERCA2a) is a key regulator in cardiac cells and its function and expression are decreased in heart failure (HF). Our group has shown that SERCA2a gene transfer induces beneficial effects in HF models and in patients with severe HF. Recently, our group determined that SERCA2a is SUMOylated by small ubiquitin related modifier type 1 (SUMO1), ultimately impacting cardiac function. The expression of SUMO1 in the myocardium is significantly decreased in failing hearts and its knockdown results in severe HF. However, the mechanisms by which SUMO1 is regulated remain unknown.
Methods/Results: To identify potential miRNAs targeting SUMO1, we first generated 19 candidates by sequence based prediction. Among 19 candidates, miR-146a was upregulated concurrently with SUMO1 expression in murine models of HF and in human HF ventricular tissues. Using a luciferase reporter assay, we confirmed that miR-146a targets the SUMO1 3’-untranslated region. Overexpression of miR-146a suppressed the expression of SUMO1 in mRNA and adversely affected the calcium transient in cardiomyocytes. Conversely, transfection of ‘decoy’ genes which has tandem complementary sequences of miR-146a increased both mRNA and protein of SUMO1. Introduction of miR-146a ‘decoy’ increased sumoylation of SERCA2a in cardiomyocytes.
Conclusion: These findings provide new insight into the regulation of SUMO1 in cardiomyocytes. It implies modulation of miR-146a is a potential target in HF therapy.
Key Words: heart failure, microRNA, SUMO, SERCA2a
Article Information
vol. 130 no. Suppl 2 A16977
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Jae Gyun Oh1;
- Ahyoung Lee1;
- Dongtak Jeong1;
- Alessia Baccarini2;
- Brian D Brown2;
- Changwon Kho1;
- Roger J Hajjar1
- 1Dept of Cardiology, Icahn Sch of Medicine at Mount Sinai, New York, NY
- 2Dept of Genetics and Genomic Sciences, Icahn Sch of Medicine at Mount Sinai, New York, NY
Abstract 16332: MicroRNA Mediated Proliferation of Human iPS Derived Cardiomyocytes for Cardiac Repair
Shazia Durrani, Raghav Pandey, Shujia Jiang, Rafeeq P Ahmed
Circulation. 2014;130:A16332
Abstract
Background: An innovative approach to generate cells for cardiac regeneration is to reprogram somatic cells into iPS and differentiate them into cardiomyocytes (CMs). Recent studies have shown micro-RNAs (miRs) can induce proliferation of neonatal rat and mouse cardiomyocytes. Here we report that overexpression of specific miRs can induce proliferation of human iPS (HiPS) derived CM and improve cardiac regeneration and function.
Methods and Results: Studies by Eulalio et. al, in 2012 identified a list of miRs that induced proliferation in neonatal mammalian cardiomyocytes. Here we transiently transfected human iPS derived CMs with miR-cel-67 (control miR), miR-1273, miR-210, and miR-1825 and administered EDU (5μM) to label proliferating cells. Five days post transfection, cells were fixed and stained for cardiomyocytes marker troponin-I, and EDU. A significant increase in proliferation was observed with miR-210 and miR-1825, while miR-1273 did not show any significant change (21.03 % and 42.97% with miR-210 and miR-1825, respectively as compared to 7.2% with miR-1273 and 6.9% with control miR-cel-67; p<.05). We selected miR-1825 for in-vivo cell transplantation experiments using immunocompromised nude mice (NOD.CB17-Prkdcscid/J). HiPS CMs were transfected with either control miR-cel-67 or miR-1825 and transplanted in mice hearts following LAD ligation. Cell control group received DMEM without cells (N= 8/group). EDU was administered IP at a concentration of 500μg/100μl per mouse, every alternate day, up to day 12 after transplantation. Four weeks after LAD ligation hearts were harvested for immuno-histology studies. Both the cell transplanted groups showed a significant reduction in infarction size when compared to DMEM control group (DMEM control 48.75+/- 5.6% vs miR-1825 HiPS CMs 33.86 +/- 4.0%; p<.05).
Conclusion: We here show a significant increase in proliferation of HiPS CMs following miR-210 and miR-1825 transduction. Our in-vivo studies show a reduction in infarction size in mice hearts that were transplanted with HiPS CMs compared to DMEM control. Collectively, this study proposes a novel approach of transplanting proliferating HiPS-CMs to acheive cardiac regeneration following ischemic injury.
Article Information
vol. 130 no. Suppl 2 A16332
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Shazia Durrani;
- Raghav Pandey;
- Shujia Jiang;
- Rafeeq P Ahmed
- Pathology/ Regenerative medicine, Univ of Cincinnati, Sch of Medicine, Cincinnati, OH
Abstract 20581: Renin Angiotensin Aldosterone System Genotype in Adults Late After Fontan Palliation
Luke J Burchill, Andrew N Redington, Candice K Silversides, Heather J Ross, Seema Mital, Laura Jimenez-Juan, Erwin N Oechslin, Cameron Slorach, Luc Mertens, Rachel M Wald
Circulation. 2014;130:A20581
Abstract
Introduction: Adults with single ventricle physiology palliated with a Fontan circulation experience high morbidity and mortality due to circulatory failure. Renin-angiotensin-aldosterone (RAAS) genotype contributes to increased cardiovascular morbidity and mortality in various forms of acquired heart failure. This study evaluated the prognostic value of RAAS genotype evaluation and serum brain natriuretic peptide in a contemporary cohort of adults following Fontan palliation.
Hypothesis: RAAS genotype would be associated with increased ventricular mass late after the Fontan operation.
Methods: Single-centre prospective study of adults (n=106) with single ventricle physiology following Fontan palliation (median age 27±9 years). Patients were genotyped for 5 pro-hypertrophic RAAS gene polymorphisms. Serum BNP levels, ventricular mass and function, and clinical events were compared between those with ≥2 homozygous risk genotypes (‘high-risk”, n=31) versus those with ≤1 homozygous risk genotypes (‘low risk”, n=75).
Results: High-risk genotype was associated with impaired ventricular compliance and higher serum BNP levels. There was no association between RAAS genotype, ventricular mass, systolic function, and/or adverse cardiovascular events. Late Fontan failure occurred in 20% of patients. Serum BNP emerged as an independent predictor of late Fontan failure [HR 1.11 (CI 1.01 – 1.23) for 50 unit increase in BNP, p=0.04] and death [HR 1.25 (CI 1.07 – 1.47) for each 50 unit increase in BNP, p=0.006].
Conclusions: Late Fontan failure is common among adults with single ventricle physiology. Contrary to our hypothesis, RAAS genotype is not associated with increased ventricular mass late after the Fontan operation. However, high-risk genotype is associated with impaired ventricular compliance and neurohormonal activation with higher serum BNP being of prognostic significance in this population.
Article Information
vol. 130 no. Suppl 2 A20581
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Luke J Burchill1;
- Andrew N Redington2;
- Candice K Silversides3;
- Heather J Ross4;
- Seema Mital2;
- Laura Jimenez-Juan5;
- Erwin N Oechslin3;
- Cameron Slorach6;
- Luc Mertens6;
- Rachel M Wald3
- 1Adult Congenital Heart Disease, Oregon Health Science Univ, Portland, OR
- 2Pediatric Cardiology, Hosp for Sick Children, Toronto, Canada
- 3Adult Congenital Heart Disease, Toronto Congenital Cardiac Cntr for Adults, Toronto, Canada
- 4Advanced Heart Failure and Heart Transplantation, Toronto General Hosp, Toronto, Canada
- 5Dept of Med Imaging, Sunnybrook Health Sciences, Toronto, Canada
- 6Echocardiography, Hosp for Sick Children, Toronto, Canada
Abstract 16976: A Mouse Model of Human Congenital Heart Disease: High Incidence of Diverse Cardiac Anomalies, Ventricular Noncompaction and Progressive Atrioventricular Block by Heterozygous Nkx2-5 Homeodomain Missense Mutation
Michelle Melanson, Hassan Ashraf, Rajib Chowdhury, Michael Silberbach, Hiroko Wakimoto, D.Woodrow Benson, Robert Anderson, Hideko Kasahara
Circulation. 2014;130:A16976
Abstract
Introduction: Heterozygous human NKX2-5 mutations are highly penetrant and associated with varied congenital heart defects (CHD) as well as progressive postnatal atrioventricular (AV) block, usually requiring pacemaker implantation. Heterozygous knockout of murine Nkx2-5, in contrast, manifests less profound CHD with low disease penetrance.
Hypothesis: We hypothesized that heterozygous knock-in having a disease-causing missense mutation rather than heterozygous knockout will mimic the phenotype of human patients.
Methods: Homeodomain (HD, DNA binding domain) missense mutations are the most frequently reported type of human NKX2-5 mutation, thus we replicated this genetic defect in a murine knock-in model (Arg52Gly) in a 129/Sv genetic background. Mouse phenotype was analyzed by histopathology (serial tissue sectioning, immunostaining), surface and telemetry ECG, and in vivo electrophysiology and compared to control Nkx2-5+/+ or Nkx2-5+/- mice at different postnatal stages from neonatal to 17 months of age.
Results: All heterozygous neonatal Nkx2-5+/R52G mice demonstrated a prominent trabecular layer in the ventricular wall, so-called noncompaction, along with diverse CHD, including ventricular septal defects (82%, n=17), atrioventricular septal defects (18%), and tricuspid valve anomalies (47%) such as Ebstein’s malformation. In addition, P10 Nkx2-5+/R52G mice demonstrated atrial septal anomalies, with significant increase in the size of the inter-atrial communication and fossa ovalis, and decrease in the length of the flap valve compared to control Nkx2-5+/+ or Nkx2-5+/- mice.
PR-prolongation (1st degree AV block) was present at 7 and 17 months of age but not at P1 and 4 weeks of age in Nkx2-5+/R52G versus control Nkx2-5+/+ mice. Advanced AV block was occasionally demonstrated in Nkx2-5+/R52G. In vivo electrophysiology studies showed that AV nodal function and right ventricular effective refractory period were impaired in Nkx2-5+/R52G mice, while sinus nodal function was not affected.
Conclusion: The effects of the human heterozygous missense mutations in NKX2-5 HD with highly penetrant, pleiotropic cardiac effects and progressive AV block was replicated in mice by introducing the missense mutation instead of knock-out.
Article Information
vol. 130 no. Suppl 2 A16976
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Michelle Melanson1;
- Hassan Ashraf1;
- Rajib Chowdhury1;
- Michael Silberbach2;
- Hiroko Wakimoto3;
- D.Woodrow Benson4;
- Robert Anderson5;
- Hideko Kasahara1
- 1Dept of Physiology and Functional Genomics, Univ of Florida College of Medicine, Gainesville, FL
- 2Dept of Pediatric Cardiology, Oregon Health Science Univ, Portland, OR
- 3Dept of Genetics, Harvard Med Sch, Boston, MA
- 4Dept of Pediatrics, Med College of Wisconsin, Milwaukee, WI
- 5Institute of Genetic Medicine, Newcastle Univ, London, United Kingdom
Abstract 16160: Common Autosomal Variants are Associated With Bicuspid Aortic Valve in Turner Syndrome
Siddharth Prakash, Michael Silberbach, Federico Asch, Giuseppe Limongelli, Hector Michelena, Dongchuan Guo, Cheryl Maslen, Carolyn A Bondy, Dianna M Milewicz
Circulation. 2014;130:A16160
Abstract
Introduction: The prevalence of bicuspid aortic valves (BAV) is enriched thirty-fold in women with Turner Syndrome (TS) in comparison with the general population.
Hypothesis: Common autosomal variants influence the development of BAV in TS women, who may be uniquely sensitized to these variants by the loss of one X chromosome. We sought to identify autosomal BAV susceptibility genes in a cohort of TS women (average age 30 years, 38% BAV, 18% coarctation).
Methods: A total of 106 TS women of European ancestry with BAV and 173 TS women with tricuspid aortic valves were genotyped on Illumina Omni-Express arrays. Valve phenotypes were determined by independent review of echocardiograms from the enrolling sites. Tests of association were performed using logistic regression without adjustment for covariates and were summarized in a meta-analysis.
Results: Xp dosage was inversely and quantitatively associated with BAV status (P=0.02). Large, recurrent copy number variants in 1p36.13, 3q29, 8p23.1 and 9p24.3 were significantly enriched in BAV cases. After exclusion of 26 outlier samples in multidimensional scaling analysis, there was no significant genomic inflation (lambda= 1.02). The strongest genome-wide association signals were observed in 1p36.23, 3q23, 12q21.2, 18q21 and 22q13.31, and did not overlap with previously reported loci for BAV. A total of 13 SNPs in 18q21 were positively associated with BAV (OR=2.5-4.3) with a minimum P value of 1×10-7. Replication of these regions in independent groups of cases is ongoing.
Conclusion: Our results demonstrate that autosomal variants with large magnitudes of effect contribute to BAV in TS women, confirming our hypothesis, and provide evidence for gene-gene interactions in BAV formation.
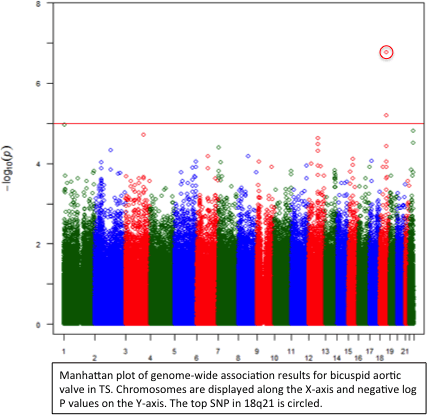
Article Information
vol. 130 no. Suppl 2 A16160
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Siddharth Prakash1;
- Michael Silberbach2;
- Federico Asch3;
- Giuseppe Limongelli4;
- Hector Michelena5,
- GenTAC Registry Consortium;
- Dongchuan Guo1;
- Cheryl Maslen6;
- Carolyn A Bondy7;
- Dianna M Milewicz1
- 1Internal Medicine, The Univ of Texas Health Science Cntr at Houston, Houston, TX
- 2Pediatrics, Oregon Health & Science Univ, Portland, OR
- 3Internal Medicine, MedStar Health Rsch Institute, Washington, DC
- 4Cardiology, Second Univ of Naples, Naples, Italy
- 5Cardiovascular Diseases, Mayo Clinic, Rochester, MN
- 6Molecular & Med Genetics, Oregon Health & Science Univ, Portland, OR
- 7Genetics of Puberty and Reproduction, National Institute of Child Health and Human Development, Bethesda, MD
Abstract 13269: Reduced Expression of scaRNAs Disrupts Spliceosome Function and Heart Development in Zebrafish and Infants with Tetralogy of Fallot
Douglas C Bittel, Prakash Patil, Tamayo Uechi, Nataliya Kibiryeva, Jennifer Marshall, Mike Artman, James E O’Brien, Naoya Kenmochi
Circulation. 2014;130:A13269
Abstract
The splicing of messenger RNA plays a fundamental role in regulating vertebrate development and differentiation. Although it is well established that alternative splicing (AS) plays an important role in regulating mammalian heart development, a clear link between misregulated splicing and congenital heart defects has not been shown. We recently reported that more than 50% of genes associated with heart development had significant changes in splice forms in the right ventricle of infants with tetralogy of Fallot (TOF; 14M/7F; all less than 1 yr old). Moreover, there was a significant decrease (30-50%, p<0.05) in the level of 12 scaRNAs. scaRNAs are members of the large family of noncoding small RNAs that are responsible for biochemical modification of specific nucleotides in spliceosomal and ribosomal RNAs. These 12 scaRNAs target two spliceosomal RNAs, U2 and U6. We used primary cells derived from the RV of infants with TOF to show a direct link between scaRNA levels and splice isoforms of several key genes regulating human heart development (e.g., GATA4, NOTCH2, DAAM1, DICER1, MBNL1 and 2). In addition, using available RNA-Seq data, we provide evidence that during zebrafish development, there are dynamic oscillations in scaRNAs and splice isoforms of genes that regulate heart development. We knocked down the expression of two scaRNAs; ACA35 (Scarna1) and U94 (Snord94), in zebrafish and saw a corresponding disruption of heart development. Importantly, there was an accompanying alteration in the ratios of splice isoforms of key cardiac regulatory genes. Based on these combined results, we propose that scaRNAs directly regulate the proficiency of the spliceosome by controlling spliceosomal RNA maturation. This in turn contributes to splice isoform dynamic equilibrium and ultimately heart development. These results are consistent with a failure of normal temporal and spatial splicing patterns during early embryonic development, leading to a breakdown in communication between the first and second heart fields, resulting in conotruncal misalignment and TOF. Our findings represent a new paradigm for understanding congenital cardiac malformations.
Article Information
vol. 130 no. Suppl 2 A13269
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Douglas C Bittel1;
- Prakash Patil2;
- Tamayo Uechi2;
- Nataliya Kibiryeva1;
- Jennifer Marshall1;
- Mike Artman3;
- James E O’Brien1;
- Naoya Kenmochi2
- 1Ward Family Heart Cntr, Children’s Mercy Hosps and Clinics, Kansas City, MO
- 2Frontier Science Rsch Cntr, Univ of Miyazaki, Miyazaki, Japan
- 3Pediatrics, Children’s Mercy Hosps and Clinics, Kansas City, MO
Abstract 11634: Linkage of Bicuspid Aortic Valve to a Chromosome 18q Locus is Associated With Aortic Dilation
Lisa J Martin, Xue Zhang, D Woodrow Benson
Circulation. 2014;130:A11634
Abstract
Introduction: Bicuspid aortic valve (BAV), a congenital valve defect, is the most common cardiovascular malformation (CVM), occurring in 1-2% of humans. BAV is a clinically significant problem as it is often associated with aortic valve disease and aortic (Ao) dilation. However, the clinical impact of BAV varies greatly; some individuals require surgical intervention in childhood, others as adults, and some have minimal or no clinical implications. Little is understood of clinical factors contributing to the clinical course. Previously, we showed BAV is determined largely by genetic effects and identified a linkage signal on chromosome (chr) 18q.
Hypothesis: To test the hypothesis that genetic variation may help explain differences in clinical course, we studied families well phenotyped by echocardiography, ascertained by probands with either BAV or hypoplastic left heart syndrome and enriched for BAV.
Methods: As Ao dimensions have been shown to be increased in individuals with BAV, we used linear regression to compare dimensions in individuals from families linking to chr 18 to those not linking to chr 18. Ao dimensions normalized based on age, sex and BSA using the mean and the standard deviation from the non BAV participants, included ascending aorta (AoA), descending aorta (AoD), aortic root (AoR), sinotubular junction (STJ), aortic valve annulus (AoVA) and transverse aorta (AoT). To account for multiple testing, a threshold of 0.008 was required for significance.
Results: A total of 1143 individuals (175 with BAV) from 209 families entered the analysis. Of these, 278 individuals from 27 families linked to chr 18. We found that BAV was associated with all Ao measures except AoD (p < 0.0001 for all). Importantly, AoR and AoT exhibited significant interactions (p = 0.0013 or 0.0033), respectively) between BAV and chr18 linkage suggesting that individuals with BAV from these families had the highest measures of AoR and AoT. Further, only AoR was significantly associated with chr18 as a main effect (p = 0.0004), suggesting that AoR is elevated in chr18 families even without underlying BAV.
Conclusions: Taken together, these results suggest that the complex genetic etiology of BAV may contribute to the clinical course as manifest by Ao dilation.
Article Information
vol. 130 no. Suppl 2 A11634
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Pediatrics, Cincinnati Children’s Hosp Med Cntr, Cincinnati, OH
- 2Pediatrics, Med College of Wisconsin, Milwaukee, WI
Abstract 17246: Predictors for Listing for Transplant in Infants with Hypoplastic Left Heart Syndrome : an analysis of the Single Ventricle Reconstruction Trial
Aparna Kulkarni, Richard Neugebauer, Jacqueline M Lamour, Daphne T Hsu
Circulation. 2014;130:A17246
Abstract
Background: Infants with Hypoplastic Left Heart Syndrome (HLHS) following the Norwood procedure have the worst survival among infant heart transplant (HT) recipients. The Single Ventricle Reconstruction (SVR) trial collected vital status and HT data in 920 eligible patients (pts) and prospective preoperative, operative and post-operative data in 554 randomized pts. This trial provides a unique opportunity to evaluate predictors of the need for HT in HLHS infants.
Methods: The public use SVR database was used to analyze factors associated with listing for HT and mortality on the wait list and in HT pts. Pts listed for HT were compared with those who survived without listing.
Results: Among 920 eligible pts, 41 were listed for HT and follow up data was available in 33/554 pts. 25 pts underwent HT. The median age at listing was 135 days (6 [[Unable to Display Character: –]] 713 days). All pts listed were status 1A, except for 2 at status 7 and 1 at status 2. Mean RV fractional area change at birth was significantly lower in the listed group (29±8% vs.35±9, p<0.05). ECMO/ CPR, and need for pacemaker were also more common in listed pts compared to the unlisted group. Gestational age, anatomic diagnosis of aortic atresia, AV valve insufficiency and the number of catheterization interventions were not risk factors for HT listing. The median time from listing to HT was 47 days (2 – 340 days). ECMO or VAD support was used in 10 pts prior to HT. Wait list mortality was 29% with a median time to death of 134 days (10 [[Unable to Display Character: –]] 707 days). Causes of death on the wait list included cardiac (8 pts), renal (1 pt), surgical (1 pt) and other (2pts). Mortality after HT was 36% with a median time to death of 197 days (48-658 days). Overall mortality after listing, including after HT, was 51%; younger age at listing was associated with increased mortality (292 vs 149 days, p<0.04).
Conclusion: HT as a rescue procedure for HLHS in the first year of life carries a significant risk of mortality.
Article Information
vol. 130 no. Suppl 2 A17246
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Pediatrics, Children’s Hosp at Montefiore, Bronx, NY
- 2Pediatrics, Columbia Mailman Sch of Public Health, New York, NY
Abstract 16801: Improved Transplant-Free Survival of Children With Dilated Cardiomyopathy: Analysis of Two Decades From the Pediatric Cardiomyopathy Registry
Rakesh K Singh, Charles Canter, Ling Shi, Steven D Colan, Debra A Dodd, Melanie D Everitt, John L Jefferies, Paul F Kantor, Minmin Lu, Elfriede Pahl, Joseph Rossano, Jeffrey A Towbin, James D Wilkinson, Steven E Lipshultz
Circulation. 2014;130:A16801
Abstract
Background: Previous studies of pediatric patients with dilated cardiomyopathy (DCM) have suggested improvement in survival has been due to utilization of heart transplantation. We sought to characterize the changing clinical characteristics and outcomes of pediatric patients with DCM phenotype over the past two decades.
Methods: Longitudinal data from 1953 children diagnosed with DCM in the NHLBI Pediatric Cardiomyopathy Registry (PCMR) from 1990-2009 were divided into two cohorts based upon year of diagnosis: “Era 1” (1990-1999, n=1199) and “Era 2” (2000-2009, n=754). Clinical information at diagnosis and incidence of transplant and death without transplant were analyzed by DCM etiology and era. Competing risks methodology was used to estimate the cumulative incidence of death or transplant by era.
Results: The majority of patients in both groups had idiopathic DCM (71 vs. 71%, p=0.96). Median age (1.6 vs. 1.7 yrs, p=0.45), left ventricular end diastolic Z scores (+4.2 vs. +4.2, p=0.84) and fractional shortening (16 vs. 17%, p=0.28) at diagnosis were similar. Era 2 patients were more likely to be treated with ACE inhibitors (71 vs. 62%, p=0.004), beta-blockers (24 vs. 6%, p<0.001), and diuretics (89 vs. 84%, p=0.002). A total of 291 patients (15%) died without transplantation, with a median time from diagnosis to death of 0.4 years. Cox regression modeling demonstrated Era 1 was associated with a higher rate of death (HR=1.5, 95% CI=1.2-2.0, p=0.002) but not transplant (0.9, 95% CI=0.7-1.1, p=0.25) when controlling for etiology group. Competing risks estimate (Figure) showed that Era 1 was associated with a higher death rate (p<0.001), while heart transplant rate was not significantly different by era (p=0.068).
Conclusions: Children with DCM phenotype have improved survival in the more recent era. This appears to be associated with factors other than the availability of transplantation, which was equally prevalent in both eras.
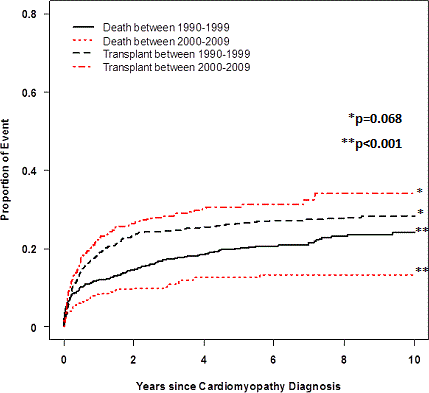
Article Information
vol. 130 no. Suppl 2 A16801
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Rakesh K Singh1;
- Charles Canter2;
- Ling Shi3;
- Steven D Colan4;
- Debra A Dodd5;
- Melanie D Everitt6;
- John L Jefferies7;
- Paul F Kantor8;
- Minmin Lu3;
- Elfriede Pahl9;
- Joseph Rossano10;
- Jeffrey A Towbin11;
- James D Wilkinson12;
- Steven E Lipshultz12,
- for the PCMR Investigators
- 1Pediatric Cardiology, Univ of California San Diego, Rady Children’s Hosp, San Diego, CA
- 2Pediatric Cardiology, Washington Univ Sch of Medicine, St. Louis, MO
- 3Statistics, New England Rsch Institutes, Watertown, MA
- 4Pediatric Cardiology, Boston Children’s Hosp, Boston, MA
- 5Pediatric Cardiology, Vanderbilt Univ, Monroe Carell Jr. Children’s Hosp, Nashville, TN
- 6Pediatric Cardiology, Univ of Utah Sch of Medicine, Primary Children’s Med Cntr, Salt Lake City, UT
- 7Pediatric Cardiology, The Heart Institute, Cincinnati Children’s Hosp Med Cntr, Cincinnati, OH
- 8Pediatric Cardiology, Univ of Alberta, Stollery Children’s Hosp, Edmonton, Canada
- 9Pediatric Cardiology, Ann and Robert H. Lurie Children’s Hosp of Chicago, Chicago, IL
- 10Pediatric Cardiology, Univ of Pennsylvania, The Children’s Hosp of Philadelphia, Philadelphia, PA
- 11Pediatric Cardiology, The Heart Institute, Cincinnati Children’s Hosp Med Cntr, Cincinnati, OH
- 12Pediatrics, Wayne State Univ Sch of Medicine, Detroit, MI
Abstract 16661: Mutation Type and Number Influence Severity of Pediatric Hypertrophic Cardiomyopathy Phenotype
Jacob Mathew, Laura Zahavich, Judith Wilson, Lee Benson, Sarah Bowdin, Seema Mital
Circulation. 2014;130:A16661
Abstract
Introduction: Improved clinical genetic testing platforms for hypertrophic cardiomyopathy (HCM) have increased detection of pathogenic lesions and of variants of unknown significance (VUS). We assessed the association of mutation type and number with severity and outcomes in pediatric HCM.
Methods: All pediatric patients at our institution who were mutation-positive for HCM genes on clinical panel or mutation-specific testing between 2000-14 were included. Patients with pathogenic variants or VUS were further analyzed. Primary outcomes included freedom from major adverse cardiac events (MACE), and severity of LV hypertrophy (LVH) during follow-up. Severe LVH was defined as a septal thickness z-score > +5. Freedom from events between different genotype groups was analyzed by the Kaplan-Meier method and Log-Rank test using STATA v12 (STATACorp, TX).
Results: Pathogenic variants or VUS were identified in 79 patients; 50 were male, 62 were familial and 41 were phenotype-negative at presentation. Mutation frequency was highest in MYBPC3 (n=42), and MYH7 (n=30), with 7 patients harboring mutations in other known HCM genes. Thirty four MACE occurred in 27 patients (9 myectomies, 20 ICDs, 3 cardiac arrests, 1 transplant, 1 death). Patients carrying mutations in MYH7 had lower freedom from MACE (HR = 2.42, p= 0.028, fig. 1) and earlier onset of severe LV hypertrophy (HR = 2.11, p=0.025). Ten patients harbored >1 lesion in one or more HCM genes (3 Pathogenic, 7 VUS). Patients with compound mutations had lower freedom from MACE compared to those with a single mutation (HR 2.54, p=0.034), which was due to lower freedom from ICD insertion (HR 4.34, p=0.001).
Conclusions: Mutations in MYH7 and occurrence of multiple mutations in one/more HCM genes was associated with higher risk of severe septal hypertrophy and MACE during follow-up in pediatric HCM patients. This suggests that secondary mutations or VUS in HCM genes may inform prognosis even in those with a known pathogenic mutation.
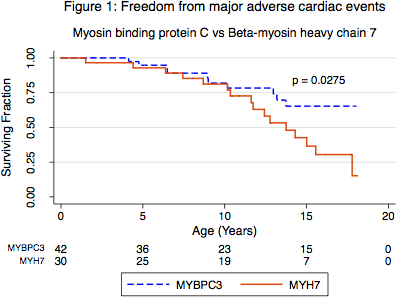
Article Information
vol. 130 no. Suppl 2 A16661
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Jacob Mathew;
- Laura Zahavich;
- Judith Wilson;
- Lee Benson;
- Sarah Bowdin;
- Seema Mital
- Cardiology, The Hosp For Sick Children, Toronto, Canada
Abstract 15622: Cost-Effectiveness of Pediatric Heart Transplantation Across a Positive Crossmatch
Brian Feingold, Steven A Webber, Cindy L Bryce, Heather E Tomko, Seo Y Park, William T Mahle, Kenneth J Smith
Circulation. 2014;130:A15622
Abstract
Introduction: Allosensitized children listed with a requirement for a negative prospective crossmatch (XM) have a high risk of death awaiting heart transplantation (HTx). Previously we found that acceptance of the first suitable organ offer for these patients, regardless of the possibility of a +XM, results in a survival benefit at all times after listing, including post-HTx. The cost-effectiveness of this strategy is unknown.
Methods: We used a Markov-state transition model with a 10 yr time horizon to compare survival, costs, and utility (i.e. quality of life) for 2 waitlist strategies for sensitized candidates: requiring a negative prospective XM (WAIT) vs. accepting the first suitable organ offer (TAKE). Model data were derived from OPTN status 1A pediatric HTx listings from 1999-2009, the PHTS and HCUP KIDS databases, and other published sources. We assumed no possibility of a +XM in the wait strategy and that the probability of a +XM in the take strategy was equal to the pre-transplant PRA.
Results: At base case, TAKE was dominant; it cost less ($122,856) and gained more (1.04) quality-adjusted life-years (QALYs) than WAIT. In sensitivity analyses varying all model parameters individually over clinically plausible ranges, TAKE remained dominant or favored (using a $100,000/QALY cost-effectiveness threshold) except when the probability of HTx for TAKE was <55% over 2 years (base case value 67%). After adjustment of the model so that waitlist probabilities of death and delisting were equal in both strategies (while maintaining the lower probability of HTx associated with WAIT), TAKE remained dominant. WAIT was no longer dominated if mortality after HTx across a +XM was >30%/year (equivalent to median post-HTx survival of <3 yrs); yet even at the extreme assumption of 100% 1-year mortality after HTx across a +XM for TAKE, the wait strategy was not cost effective ($350,097/QALY).
CONCLUSIONS: Among sensitized status 1A pediatric HTx candidates, we found that taking the first suitable organ offer is less costly and results in greater survival than awaiting HTx across a negative prospective XM. This suggests that HTx should not be denied based on sensitization status alone.
Article Information
vol. 130 no. Suppl 2 A15622
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Brian Feingold1;
- Steven A Webber2;
- Cindy L Bryce3;
- Heather E Tomko3;
- Seo Y Park4;
- William T Mahle5;
- Kenneth J Smith4
- 1Pediatric Cardiology, Children’s Hosp of Pittsburgh of UPMC, Pittsburgh, PA
- 2Pediatrics, Vanderbilt Univ, Nashville, TN
- 3Sch of Public Health, Univ of Pittsburgh, Pittsburgh, PA
- 4Medicine, Univ of Pittsburgh, Pittsburgh, PA
- 5Pediatrics, Emory Univ Sch of Medicine, Atlanta, GA
Abstract 11760: The Outcomes of Children Implanted With Ventricular Assist Devices in the US: First Analysis of the Pediatric Interagency Registry for Mechanical Circulatory Support (PediMACS)
Elizabeth D Blume, David N Rosenthal, Joseph W Rossano, J. T Baldwin, Pirooz Eghtesady, David L Morales, Ryan S Cantor, Jennifer Conway, Christopher S Almond, David C Naftel, James K Kirklin
Circulation. 2014;130:A11760
Abstract
BACKGROUND: Use of pediatric mechanical circulatory support to decrease mortality has expanded dramatically. A national account of the use of FDA-approved devices in children is essential to understanding outcomes, refining patient selection, and improving quality of care.
METHODS: PediMACs is an NIH-supported national registry for FDA-approved temporary and durable VAD usage in patients < 18 years of age. Between the launch in Sept 2012 and March 2014, 32 US hospitals enrolled patients. This first report of data from this registry analyzed pre-implant patient characteristics, survival using competing outcomes, and adverse events.
RESULTS: One hundred and thirty pediatric pts underwent 153 VAD implantations. Etiology of heart disease included 87 (67%) pts with cardiomyopathy, 26 (20%) with congenital heart disease. Twenty pts transitioned from ECMO and 37 had prior cardiac surgery. Most pts were Intermacs level 1 (28%) and level 2 (55%) at implant, with 12% Intermacs level 3. There were 26 temporary devices. Of the durable devices, 65 (51%) were pulsatile and 62 (49%) continuous flow (CF) (compared to adult Intermacs implants: 4% pulsatile, 96% CF). Age at first implant included 22 pts (17%) < 1 yr of age, 23 (18%) 1-5 yrs, 26 (20%) 6-10 yrs and 59 (45%) 10-18 yrs. Patients were supported with LVAD alone in 99 (76%), BiVAD in 24 (18%) and total artificial heart in 2 (2%) and represented 484 months of follow-up. The 104 patients receiving primary durable devices had an actuarial survival of 90% at 6 mos. Competing risk analysis at 6 mos revealed 54% of pts transplanted, 37% alive on support, 8 % died and 1 % recovery. In the overall cohort, there were 8 early (<1mo) and 5 late deaths. Reported serious adverse events included infection (42), bleeding (40), device malfunction (37) and neurologic dysfunction (33).
CONCLUSIONS: Pedimacs constitutes the largest single data repository with detailed information of pediatric pts implanted with all types of VADs. Initial data show a higher rate of use of pulsatile devices as compared to contemporaneous adult data. Survival rates are excellent despite varying patient characteristics and pump types. With further data collection, analysis of patient risk factors critical to improving outcomes will be possible.
Article Information
vol. 130 no. Suppl 2 A11760
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Elizabeth D Blume1;
- David N Rosenthal2;
- Joseph W Rossano3;
- J. T Baldwin4;
- Pirooz Eghtesady5;
- David L Morales6;
- Ryan S Cantor7;
- Jennifer Conway8;
- Christopher S Almond9;
- David C Naftel7;
- James K Kirklin7
- 1Cardiology, Boston Children’s Hosp, Boston, MA
- 2Dept of Pediatrics, Stanford Univ, Palo Alto, CA
- 3Cardiology, The Children’s Hosp of Philadelphia, Philadelphia, PA
- 4Div of Cardiovascular Sciences, National Heart, Lung, and Blood Institute, Bethesda, MD
- 5Surgery, Washington Univ Sch of Medicine, St. Louis, MO
- 6Cardiothoracic Surgery, Cincinnati Children’s Hosp Med Cntr, Cincinnati, OH
- 7Dept of Surgery, Univ of Alabama at Birmingham, Birmingham, AL
- 8Pediatric Cardiology, Stollery Children’s Hosp, Univ of Alberta, Edmonton, Canada
- 9Pediatrics (Cardiology), Stanford Univ, Palo Alto, CA
Abstract 19684: Abnormal Aortic Wall Properties in Children with Isolated Bicuspid Aortic Valve
Shobha S Natarajan, Andrew C Glatz, Elizabeth Goldmuntz, Meryl S Cohen
Circulation. 2014;130:A19684
Abstract
Introduction: Abnormal aortic wall properties have been reported in patients with isolated bicuspid aortic valve (IBAV) even in the absence of significant aortic stenosis or regurgitation.
Hypothesis: We sought to assess aortic distensibility (DIS) and stiffness index (SI) in children with IBAV compared to age group-matched subjects with normal tricuspid aortic valves (TAV) and to determine whether these abnormalities in the aortic wall properties correlate with bicuspid valve morphology or left ventricular systolic or diastolic function.
Methods: Children ages 8-18 years with an IBAV and age group-matched controls with a TAV were prospectively enrolled. Subjects with greater than mild stenosis or mild regurgitation were excluded. Using echo, aortic valve morphology, aortic root (AoR) and ascending aorta (AAo) diameters and z-scores were determined. Left ventricular shortening fraction (LVSF), DIS and SI were measured using M-mode echo. Diastolic function was determined using mitral valve septal E/Ea. Blood pressure (BP) was measured at the time of echo.
Results: Nineteen had IBAV and 17 had TAV. There were no significant differences in age, weight, height or BP between the two groups. In the IBAV group, 11 had right-left type (R/L) and 8 had right-non type (R/N). There was no significant difference in AoR z-scores between groups. The IBAV group had larger AAo z-scores (2.48±1.9 vs. -0.02±0.98, p<0.0001), decreased DIS (9.6±4 vs. 12.3±3.1 cm2 dynes-1 x 10-6, p<0.05) and increased SI (21.4±9.2 vs. 14.4±3.8, p=0.007) compared to the TAV group. There were no differences in these variables between the R/L or R/N subgroups. No correlation was seen between aortic wall properties and ventricular function in the IBAV group. By multivariate regression, presence of an IBAV (coefficient = -2.4, p=0.03), LVSF (coefficient = -0.35, p=0.01) and age-adjusted systolic BP (coefficient = -0.13, p=0.03) were independently associated with DIS. Similarly, presence of an IBAV (coefficient = 6.7, p=0.005) and age (coefficient=0.85, p=0.02) were independently associated with SI.
Conclusions: Children with IBAV have decreased DIS and SI even without hemodynamic abnormalities. Long-term studies to determine the impact of these findings on cardiovascular risk are needed.
Article Information
vol. 130 no. Suppl 2 A19684
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Shobha S Natarajan;
- Andrew C Glatz;
- Elizabeth Goldmuntz;
- Meryl S Cohen
- Cardiology, The Children’s Hosp of Philadelphia, Philadelphia, PA
Abstract 19743: Assessment of Myocardial Mechanics Utilizing Speckle-Tracking Echocardiography in a Swine Model of Right Ventricular Pressure-Overload
Nicholas Pietris, Brody Wehman, Osama T Siddiqui, Rachana Mishra, Grace Bigham, Sudhish Sharma, Sarah B Murthi, Sunjay Kaushal
Circulation. 2014;130:A19743
Abstract
Introduction: Quantitative assessment of right ventricular (RV) function by echocardiography remains a challenge due to the complex geometry of the RV. The objective of this study was to assess the utility of speckle-tracking echocardiography (STE) compared to conventional transthoracic two-dimensional (2DE) in a RV pressure-overload swine model after treatment with cardiac or mesenchymal stem cells.
Methods: Neonatal swine underwent pulmonary artery banding (PAB) to induce RV dysfunction. After banding, pigs received intramyocardial injection into the RV free wall with human c-kit+ cardiac stem cells (hCSCs) alone (n=5), human mesenchymal stem cells (hMSCs) alone (n=5), a combination of hCSCs/hMSCs (n=5) or placebo (phosphate-buffered saline, n=5). Standard 2DE was performed pre-operatively, post-banding and at 4 weeks. Offline blinded analysis using vendor-independent software was performed measuring longitudinal strain and strain rate from the apical 4-chamber view.
Results: The mean RV:systemic pressure ratio at baseline and post-banding was 0.34±0.04 vs. 0.76±0.05, respectively (P<0.0001). Compared to baseline, mean post-banding values for peak global longitudinal strain (pGLS) and strain rate were significantly decreased (P<0.0001 and P=0.0002, respectively). At four weeks post-banding, pGLS was higher in all cell-treated pigs relative to placebo (hMSCs, P=0.002; hCSCs, P<0.0001; hCSCs/hMSCs, P=0.014). Strain rate was higher than placebo at four weeks post-banding in the hMSCs alone and hCSCs alone groups, and approached statistical significance in the combination hMSCs/hCSCs. Fractional area of change demonstrated significant improvement in cell-treated pigs compared to placebo, however no significant changes were detected in other traditional measurements, such as TAPSE or tricuspid tissue Doppler indices.
Conclusion: In a neonatal model of RV pressure-overload, we were able to detect significant changes in underlying myocardial mechanics which conventional RV functional indices did not detect. STE may be more sensitive than traditional echocardiography in assessing RV function in neonatal pressure-overloaded lesions.
Article Information
vol. 130 no. Suppl 2 A19743
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Nicholas Pietris1;
- Brody Wehman2;
- Osama T Siddiqui2;
- Rachana Mishra2;
- Grace Bigham2;
- Sudhish Sharma2;
- Sarah B Murthi2;
- Sunjay Kaushal2
- 1Pediatrics, Univ of Maryland Med Cntr, Baltimore, MD
- 2Surgery, Univ of Maryland Med Cntr, Baltimore, MD
Abstract 19568: Hemodynamic Assessment of an Augmented Aorta: a Rapid Prototyping Technique
Kevin A Gralewski, Kevin K Whitehead, Yoav Dori
Circulation. 2014;130:A19568
Abstract
Introduction: Interest in high fidelity aortic flow phantoms remains significant even with advancements in computational fluid dynamic methods. We present a process for creating a patient-specific, compliant aortic arch and valve (AoV) along with our corresponding validation efforts.
Methods: A rendered aortic volume was created by threshold-based segmentation in Mimics (Materialise, Leuven, Belgium) and edited in 3-matic to create a 3D printed mold (Object Connex 5000, Stratasys, Edina, Minnesota) into which a polyurethane based resin (Smooth-on, Easton, Pennsylvania) was cast. The AoV was created in a similar manner and ultimately seated in the distal end of an inlet port designed to induce laminar flow. The arch, with fixed inlet, was then constrained to the correct anatomical conformation by a custom rapid prototyped chamber. An MRI-compatible pump programmed to match the patient’s flow profile managed flow of a 40% glycerin-aqueous solution. Both through-plane and 4D phase contrast velocity mapping MRI sequences were acquired and compared to the patient data with time-elapse flow streamlines calculated by GTFlow version 2.0.1 (GyroTools, Zurich Switzerland).
Results: The phantom remained robust and compliant throughout the dynamic loading occurring under pulsatile flow. Registration revealed good alignment of the phantom lumen to the segmented patient aorta. 4D flow analysis showed an unusual left-handed helical flow pattern in both the in vivo patient data and derived phantom flows. Flow measurements in the ascending and descending aorta of the model agreed within 5% of the actual patient measured flow.
Conclusions: We have demonstrated a viable method to create patient-specific flow phantoms, which closely mimic the physiological system for which they are modeled. Further studies are needed to optimize the valve anatomy and wall compliance.
Article Information
vol. 130 no. Suppl 2 A19568
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Kevin A Gralewski;
- Kevin K Whitehead;
- Yoav Dori
- Cardiology, The Children’s Hosp of Philadelphia, Philadelphia, PA
Abstract 18947: What Can We Learn from Serial Echocardiographic Evaluation of the Aorta in Tetralogy of Fallot?
Matthew J Lewis, Jonathan Ginns, Marlon Rosenbaum
Circulation. 2014;130:A18947
Abstract
Introduction: Guidelines for management of aortic root (AoR) dilation with known aortopathies recommend surgical repair when the AoR exceeds 5 cm. Patients with tetralogy of Fallot (TOF) may have an associated aortopathy, though aortic dissection is rare. No guidelines exist regarding timing of surgical intervention. We hypothesized that the rate of AoR dilation in adults with repaired TOF would be low and that an AoR > 5 cm would be associated with standard risk factors.
Methods: We performed a retrospective study of all adult TOF patients who underwent a transthoracic echocardiogram (TTE) at our Center. Clinical variables and AoR dimension at the sinus of Valsalva were determined. AoR size was treated as an absolute value and as an observed-to-expected ratio based on standard nomograms.
Results: 266 patients met inclusion criteria. On initial TTE, 91 (34%) patients had an AoR > 4.0 cm, 9 (3%) patients had an AoR > 5.0 cm, and 28 patients (11%) had an observed-to expected AoR > 1.5. Male gender, hypertension, and TOF repair at >5 years-old were associated with an AoR > 5.0 cm while only TOF repair at >5 years-old was associated with an observed-to-expected AoR > 1.5. 4 patients had aortic valve (AV) surgery. One patient with an AoR of 5.8 cm had an aortic dissection following pulmonary valve replacement.
206 patients (77%) had serial TTEs. Mean age at first echo was 35.5 years. Mean time between studies was 5.5 years, and 106 patients had a >5 year latency between TTEs. The mean rate of change for all patients was +0.4 mm/year. 72 patients (35%) had an increase in AoR size. 25 patients (12%) had an increase in AoR size ≥0.5cm. Only time >5 years between TTEs was significantly associated with an increase in AoR ≥0.5 cm (p = 0.014). There was no significant association with either rate of AoR increase or AoR increase ≥0.5 cm and gender, hypertension, degree of AR, AV surgery, right-sided arch, AoR> 5cm, time of repair, or prior pregnancy.
Conclusions: In our cohort of adult patients, AoR dilatation >5.0 cm was uncommon and associated with male gender, hypertension and late repair. The rate of AoR dilation was slow and without clear risk factor. In patients with severe AoR dilation, further study is required to identify significant risk factors for aortic events.
Article Information
vol. 130 no. Suppl 2 A18947
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Matthew J Lewis;
- Jonathan Ginns;
- Marlon Rosenbaum
- Cardiology, Columbia Univ/New York Presbyterian Hosp, New York, NY
Abstract 19021: Abnormalities of Ventricular Morphology and Compaction Are Prevalent in Adults With Coarctation of the Aorta – A Two-centre Cardiac Magnetic Resonance Imaging (CMR) Study
Preeti Choudhary, Christian Hamilton-Craig, Wendy Strugnell, Rajesh Puranik, David S Celermajer
Circulation. 2014;130:A19021
Abstract
Introduction: Coarctation of the aorta (CoA) is associated with valvular and aortic disease but abnormalities of ventricular morphology are less well understood. Echocardiographic case series suggest a high prevalence of left ventricular non-compaction (LVNC) however; this has not been clarified by cardiac magnetic resonance imaging (CMR).
Methods: CMR images of CoA adults were collated from two statewide tertiary referral centres in Australia. LVNC was defined as ratio of non-compacted to compacted myocardium of greater than 2.3 and abnormal compaction (LVAC) was defined by a ratio of 1.5-2.3 in end-diastole. Clinical information, volumetric data and mass calculations were obtained.
Results: CMR scans of 293 CoA patients were analysed (Mean age 30+/-13 years; 50% male). 140 patients (48%) had bicuspid aortic valves (BAV) and 14 had unrepaired CoA. LVAC was evident in 78 % of patients and LVNC in 15 %. The most prevalent abnormality was mid-ventricular septal trabeculation in 40 patients. Patients with LVNC had significantly higher indexed end-diastolic (97+/-24 mL/m2 vs. 119+/-29 mL/m2; p=0.006) and end-systolic volumes (58+/-19 mL/m2 vs. 47+/-16 mL/m2; p = 0.03) compared to normally compacted ventricles, irrespective of the severity of aortic valvular disease. Ejection fraction did not significantly differ between the LVAC and normally compacted patients. (EF 60+/-6% vs. abnormally compacted EF 52+/-1; p = 0.30). Age, gender or presence of BAV did not correlate with presence of NC. In a subset of 148 cases, detailed short axis LV cine stacks were performed, revealing abnormalities of the mitral valve leaflets in 10, papillary muscles in 17 and myocardial clefts in 28 patients.
Conclusions: Abnormal ventricular morphology is prevalent in CoA and should be considered in addition to the known valvular and vascular complications. LVNC correlates with increased end-diastolic volumes and may impact on ventricular function over time.
Article Information
vol. 130 no. Suppl 2 A19021
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Preeti Choudhary1;
- Christian Hamilton-Craig2;
- Wendy Strugnell2;
- Rajesh Puranik1;
- David S Celermajer1
- 1Cardiology, Univ of Sydney, Royal Prince Alfred Hosp, Sydney, Australia
- 2Cardiology, Richard Slaughter Cntr of Excellence for CVMRI, The Prince Charles Hosp, Brisbane, Australia
Abstract 18473: Rates and Risk Factors for Faster Aortic Root Growth in Pediatric Patients with Marfan Syndrome
Nitya Viswanathan, Claudia Pedroza, Shaine A Morris
Circulation. 2014;130:A18473
Abstract
Background: Marfan Syndrome (MS) is associated with progressive aortic dilation and aortic dissection. Aortic dissection is typically preceded by aortic dilation. The goal of this study is to identify factors associated with faster rates of aortic root dilation in children with MS.
Methods: Patients undergoing serial transthoracic echocardiograms (TTE) with MS were retrospectively identified from an institutional database. Those with >2 TTEs over 1 year apart and <21 years of age at first TTE were included. TTEs performed after aortic surgery were excluded. Using multivariable longitudinal linear regression analysis, sex, medication, presence of ectopia lentis, need for scoliosis surgery and infantile type of MS were evaluated for associations with rate of change in aortic root dimension and aortic root z-score over time.
Results: Of 240 patients with MS, 146 were included. Median age at first TTE was 8.1 yrs (range 0-20.9 years), median length of follow up 6.5 years (range 1.0-20.1 years), and median number of studies was 8 (range 2-25). Sixty-one percent were male. Of the 146 patients, 123 (84%) were documented to be on medical therapy: 14 angiotensin receptor blocker (ARB), 66 B-Blocker (BB), 10 prior history of both, 5 BB+ARB, 27 were in the Pediatric Heart Network medication trial, 1 ACE inhibitor. Sixteen patients underwent root replacement surgery at a median age of 14.6 years (range 1.8-24). No patients had aortic dissection. Three patients had infantile MS. All of these patients underwent root replacement at 1.8, 2 and 4 years of age. Two underwent subsequent aortic root replacement at 6 and 8 years old. Mean rate of aortic root growth in the cohort was 0.12cm/year, and mean change in z-score was 0.02/year (p=0.23 compared to expected rate of no change). The only variable associated with faster root growth was infantile MS (1.3cm/year, p<0.001; z-score change of 6.9/year, p<0.001). There was no significant difference in the rate of aortic root growth between patients who received therapy with BB vs. ARB vs. BB+ARB.
Conclusions: Children with MS did not have a significant change in aortic root z-score over time, and the only factor associated with more rapid aortic root growth was infantile type MS.
Article Information
vol. 130 no. Suppl 2 A18473
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Pediatrics, Texas Childrens Hosp, Houston, TX
- 2Pediatrics, Univ of Texas, Health Sciences Cntr at Houston, Houston, TX
- 3Pediatric Cardiology, Texas Childrens Hosp, Houston, TX
Abstract 17654: Late Wall Thickening and Calcification in Patients After Kawasaki Disease Noninvasively Detected by Dual Source Computed Tomography
Nobuyuki Tsujii, Etsuko Tsuda, Suzu Kanzaki, Jun Ishizuka, Koichirou Nakashima, Kenichi Kurosaki
Circulation. 2014;130:A17654
Abstract
Background: Late coronary wall thickening (WT) including coronary calcification indicates an irreversible change of the coronary artery after Kawasaki disease (KD), which can be detected noninvasively by Dual Source Computed Tomography (DSCT).
Purpose: We retrospectively investigated the relation between coronary artery lesion (CAL) in the acute KD and late WT detected by DSCT.
Methods: Sixty-two pts (47 males and 15 females) who had previously undergone selective coronary angiograms (CAGs) less than 100 days after the onset of KD were studied by DSCT. The age at DSCT ranged from 10 months to 36 years (median, 18 years). The interval from the onset of KD to DSCT ranged from 26 days to 34 years (median, 16 years). DSCT was performed using a SOMATOM® Definition Flash (Siemens). The maximum diameters of segments 1, 2, 3, 6, 7, and 11 were measured in the initial CAGs (Branches group; BG), and the bifurcation of the left coronary artery was also measured (LCA). WT was diagnosed by two observers. We studied the relationship between the maximum diameter of coronary artery detected by the initial CAG, the interval from the onset of KD and the appearance of WT detected by DSCT. We determined the cut-off point of coronary artery dilatation for WT in each group by using Receiver Operating Characteristic (ROC) analysis.
Results: WT in the BG and the LCA were detected in 119/326 and 24/39, respectively. The appearance of late WT in CAL of acute phase was 115/190 (61%), and that in no CAL was 4/136 (3%) (p<0.001) in the BG group. In both groups, the appearance of WT was significantly related with the initial diameters and the interval from onset of KD, respectively. The cut-off point of acute coronary dilatation for WT in BG was 4.8mm (AUC 0.86, p<0.0001), and that in LCA was 7.9 mm (AUC 0.65, p=0.0464).
Conclusions: Acute coronary dilatation more than 4.8 mm in BG and 7.9 mm in LCA can lead to late WT. DSCT was useful to detect late WT in patients after KD.
Article Information
vol. 130 no. Suppl 2 A17654
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Nobuyuki Tsujii1;
- Etsuko Tsuda1;
- Suzu Kanzaki2;
- Jun Ishizuka1;
- Koichirou Nakashima1;
- Kenichi Kurosaki1
- 1Dept of Pediatric Cardiology, National Cerebral and Cardiovascular Cntr, Osaka, Japan
- 2Dept of Radiology, National Cerebral and Cardiovascular Cntr, Osaka, Japan
Abstract 15789: Cardiac MRI With Adenosine Identifies Coronary Stenosis, Perfusion Defects, and Delayed Enhancement in Pediatric Patients With Anomalous Aortic Origin of a Coronary Artery Before and After Surgical Repair
Julie A Brothers, Timothy S Kim, Mark A Fogel, Kevin K Whitehead, Stephen A Paridon, Matthew A Harris
Circulation. 2014;130:A15789
Abstract
Background: Anomalous aortic origin of a coronary artery (AAOCA) with an interarterial course is associated with sudden cardiac death in children.
Objectives: Using cardiac MRI with adenosine, we evaluated coronary ostial stenosis, proximal coronary size, and left ventricular (LV) function in children with AAOCA.
Methods: We prospectively enrolled children 5-18 years old with interarterial AAOCA. MRIs were reviewed for coronary artery origin, proximal course, dimensions, and cardiac function. Surgery consisted of the modified unroofing procedure. We used descriptive statistics and paired t-tests to evaluate for statistical significance.
Results: Between 2/2009 and 5/2014, 24 subjects with AAOCA underwent 29 MRIs. The majority were male (N=19, 79%) with anomalous right coronary artery (AAORCA, N=20, 83%). Mean age was 12.8 years at time of initial MRI. MRI was performed an average of 7 months post-operatively in 8 subjects who underwent surgery. In all, the proximal anomalous coronary arose tangential to the aorta with an elliptical, slit-like ostium. The anomalous coronary measured smaller proximally (0.20 mm) compared to distally (0.31 mm, P=< 0.0001), and after surgical repair, the post-operative origin was significantly larger (0.36 vs. 0.21 mm, P=0.02). Other abnormalities at initial MRI included fixed inferior wall (N=1) and reversible subendocardial septal/inferior wall (N=1) perfusion defects. Post-operatively, the neo-ostium was round in 6 (see Figure), but in 2, the orifice remained elliptical. One patient had a new small mid-myocardial scar and one had dyskinetic septal wall motion. LV function was normal both before and after surgery (mean ejection fraction =68.1% vs. 67.5%, P=0.85).
Conclusions: Cardiac MRI with adenosine is an important tool for the evaluation of anomalous anatomy, myocardial function, and ischemia/injury and should be considered for the initial and, when applicable, post-operative assessment of children with AAOCA.
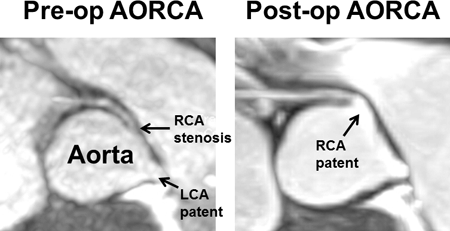
Article Information
vol. 130 no. Suppl 2 A15789
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Julie A Brothers1;
- Timothy S Kim2;
- Mark A Fogel2;
- Kevin K Whitehead2;
- Stephen A Paridon2;
- Matthew A Harris1
- 1Pediatric Cardiology, The Children’s Hosp of Philadelphia, Philadelphia, PA
- 2Cardiology, The Children’s Hosp of Philadelphia, Philadelphia, PA
Abstract 12779: Increased Diastolic Untwisting Velocity in Response to Tachycardia as Evidence of Diastolic Reserve in the Young Infant Heart: A Simultaneous Invasive and Noninvasive in vivo Swine Model
Etienne Fortin-Pellerin, Lindsay Mills, James Y Coe, Nee S Khoo, Po Y Cheung, Lisa K Hornberger
Circulation. 2014;130:A12779
Abstract
Introduction: In response to exercise, the healthy adult left ventricle (LV) augments its filling through early untwisting, creating suction even before the AV valve opens. The role of untwisting in the immature heart remains controversial. Older infants have delayed and decreased LV untwisting rates at baseline, suggesting untwisting may play less of a role in LV filling at least at rest. Although no in vivo data exists, one in vitro investigation of human infant myocardium has suggested relaxation may be augmented during tachycardia which could suggest an element of diastolic reserve. In the present study, we sought to explore the diastolic function response to atrial tachycardia in a young piglet model.
Methods: Under general anesthesia (propofol, isoflurane), 1-15 day old piglets were instrumented intravascularly with Millar high-fidelity and pacing catheters in the LV and right atrium (RA), respectively. After stabilization, invasive hemodynamic and echocardiography parameters were acquired at baseline and at 230bpm (30-40% above baseline). LV twist was analyzed off-line by speckle tracking (226 ± 55 frames/s). Subjects were their own control and paired t-tests were used for comparisons after confirmation of normal distribution for each variable. Values were expressed as mean ± SD.
Results: Eight piglets of mean age 8.6 ± 6.8 days and weight 3.6 ± 2.18kg, and baseline heart rate of 157 ± 18bpm were assessed. With tachycardia, Tau decreased from 28 ± 9ms to 23 ± 9ms (p = 0.03). Peak untwisting rate increased from -247 ± 83 to -412 ± 179 degrees/s (p = 0.04) and the change correlated with Tau (r=0.45, p=0.04). Untwisting rate during isovolumic relaxation also increased from -146 ± 74 deg/s to -335 ± 205 deg/s (p = 0.03). There was a trend towards reduction in LV end diastolic pressure from 13 ± 6 to 10 ± 5 mmHg (p = 0.067).
Conclusion: The early infant heart has the capacity to maintain normal LV filling pressures during atrial tachycardia, and this is associated with increased LV untwisting performance suggesting diastolic reserve. The boundaries of this diastolic reserve, and whether this knowledge can be exploited to augment LV filling in the critically ill infant is the subject of ongoing investigations.
Article Information
vol. 130 no. Suppl 2 A12779
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Etienne Fortin-Pellerin1;
- Lindsay Mills1;
- James Y Coe1;
- Nee S Khoo1;
- Po Y Cheung2;
- Lisa K Hornberger1
- 1Pediatric Cardiology, Stollery Children’s Hosp, Edmonton, Canada
- 2Neonatology, Stollery Children’s Hosp, Edmonton, Canada
Abstract 11138: Simplified Rheumatic Heart Disease Screening Criteria for Handheld Echocardiography
Jimmy C Lu, Craig Sable, Gregory J Ensing, Catherine C Webb, Janet Scheel, Twalib Aliku, Justin C Godown, Andrea Beaton
Circulation. 2014;130:A11138
Abstract
Background: Using 2012 World Heart Federation (WHF) criteria, standard portable echocardiography (SPE) reveals a high burden of rheumatic heart disease (RHD) in resource poor settings, but widespread screening is limited by cost and physician availability. Handheld echocardiography (HHE) may decrease costs, but WHF criteria are complicated for rapid field screening, particularly for non-physician screeners.
Objective: To determine the best simplified screening strategy for RHD detection using HHE.
Methods: HHE (GE Vscan) and SPE (GE Vivid q or i or Philips CX-50) were performed in 5 schools in Gulu, Uganda. Borderline or definite RHD cases were defined by 2012 WHF criteria on SPE images, by 2 experienced readers. HHE studies were reviewed by cardiologists blinded to SP results. As HHE lacks continuous wave Doppler, pansystolic mitral regurgitation (MR) was defined as MR on 2 consecutive frames. We evaluated single and combined HHE parameters to determine the simplified screening strategy that maximized sensitivity and specificity for case detection.
Results: In 1420 children (10.8±2.6 years old, 47% male) with HHE and SPE studies, morphologic criteria and presence of any MR by HHE had poor specificity (Table). MR jet length by HHE correlated with SPE (r=0.54, p<0.0001). Aortic insufficiency (AI) was specific but not sensitive. Combined criteria of MR jet length ≥1.5 cm, chosen from receiver operating characteristic analysis, or any AI best balanced sensitivity and specificity; pansystolic MR could be substituted for MR jet length with slightly lower sensitivity for definite RHD. With a prevalence of 4% and subsequent SPE screening of positive HHE studies, this would reduce SPE studies by 80% from a SPE-based screening strategy.
Conclusions: In resource-limited settings, HHE with simplified criteria offers reasonable sensitivity and specificity for RHD screening. Further study is needed to validate HH screening by local practitioners and long-term outcomes.
Article Information
vol. 130 no. Suppl 2 A11138
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Jimmy C Lu1;
- Craig Sable2;
- Gregory J Ensing1;
- Catherine C Webb1;
- Janet Scheel2;
- Twalib Aliku3;
- Justin C Godown1;
- Andrea Beaton2
- 1Pediatrics and Communicable Diseases, Univ of Michigan, Ann Arbor, MI
- 2Cardiology, Children’s National Health System, Washington, DC
- 3Pediatric Cardiology, Uganda Heart Institute, Kampala, Uganda
Abstract 20435: Myocardial Mechanics of the Systemic Right Ventricle in Hypoplastic Left Heart Syndrome Through the Perinatal Transition
Santokh S Dhillon, Akiko Hirose, Nee S Khoo, Lindsay Mills, Timothy Colen, Winne Savard, Po Yin Cheung, Lisa Hornberger
Circulation. 2014;130:A20435
Abstract
Background: Rapid changes occur in myocardial loading during the transition from the fetal to postnatal circulation. In healthy neonates, the LV output almost doubles within hours of delivery but decreases to levels slightly higher than the fetal LV output within the first few days. In contrast, we have found in Hypoplastic left heart syndrome (HLHS) the combined cardiac output (CO) is initially similar to that of the late gestation fetus with a gradual but progressive increase occurring within the first 3-5 days. We sought to define the functional changes of the systemic right ventricle (RV) during the perinatal transition in the neonate with HLHS.
Methods: We prospectively recruited 11 pregnancies with fetal HLHS. Echocardiograms were performed in late gestation and at 4 -12, 20-24, 44-48 hours (hrs) and 3-5 days after birth. Hemodynamics including stroke volume (SV), heart rate (HR), CO, blood pressure (BP), and RV size and fractional area change (FAC) were measured. High frame rate images were used for offline strain and strain rate (SR) analysis using speckle tracking. ANOVA with repeated measures and paired t-test for parameters and Pearson’s correlations were used.
Results: In the first 24 hrs, CO increased (p=0.03) which was accompanied by an increase in RV longitudinal strain (p=0.03). From 24 hrs to 5 days, although CO and SV continued to increase (p<0.001 for both) along with progressive increase in RV end diastolic size (p<0.002), no further increase in longitudinal strain and SR was observed. There was no change in HR, systolic BP or RV FAC between intervals. Longitudinal strain and SR correlated with CO (r=-0.44; p=0.007 & r=-0.60; p=0.0001 respectively) and SV (r=-0.46; p=0.004 & r=-0.55; p=0.0006 respectively). Longitudinal SR also correlated with RV FAC (r=-0.40; p=0.01) and RVEDV (r =0.37; p=0.02).
Conclusion: The RV in neonatal HLHS appears to compensate by initially increasing its longitudinal deformation in the first 24 hrs to meet the postnatal circulatory demands. Thereafter, subsequent increase in CO relies on increasing SV through progressive increase in ventricular preload. Further studies using deformation imaging are needed to explore RV adaptation in early transition in HLHS neonates and its implication on management and outcome.
Article Information
vol. 130 no. Suppl 2 A20435
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Santokh S Dhillon;
- Akiko Hirose;
- Nee S Khoo;
- Lindsay Mills;
- Timothy Colen;
- Winne Savard;
- Po Yin Cheung;
- Lisa Hornberger
- Fetal & Neonatal Cardiology program, Univ of Alberta, Edmonton, Canada
Abstract 19791: Echocardiography Derived Velocity Time Integral is Associated with Double Switch Outcomes in Congenitally Corrected Transposition of the Great Arteries
Shreya Moodley, Sowmya Balasubramanian, Theresa Tacy, Frank Hanley, Frandics Chan, Rajesh Punn
Circulation. 2014;130:A19791
Abstract
Introduction: Congenitally corrected transposition of the great arteries (CCTGA) is a rare form of congenital heart disease characterized by atrioventricular and ventriculoarterial discordance. Management is controversial; options include observation, physiologic repair and anatomic repair by the double switch operation (DSO). Assessment of morphologic left ventricle (LV) preparedness is key in determining DSO timing, although exact criteria are lacking. Cardiac MRI is the current standard technique to assess LV adequacy prior to DSO. The purpose of our study was to determine if echocardiographic (echo) or MRI measures of the LV predict outcomes.
Methods: A retrospective review of CCTGA patients eligible for DSO at Lucile Packard Children’s Hospital from 2000-2014 was conducted. Inclusion criteria were: 1) age <18 years, 2) adequate pre-surgical echo and MRI images and 3) clinical follow-up information available. Post-processing of echo images was performed to obtain measurements of LV structure and function. Measurements included 5/6 and 2/3 area-length ejection fraction (EF) and mass, LV eccentricity index, LV posterior wall thickness, LV tissue Doppler velocities, tricuspid valve regurgitation severity, pulmonary artery (PA) pressure gradient and PA velocity time integral (VTI). MRI measurements included LV mass and EF. Outcomes included achieving DSO and freedom from death, transplant and heart failure at last follow up.
Results: 31 patients met inclusion criteria (7.3 ± 7.8 years). Peak PA VTI correlated significantly with outcomes. Receiver operating curve analysis showed that a VTI of > 1.17cm predicted successful DSO and freedom from death, transplant and heart failure at last follow up, with a respective sensitivity of 80% and 83% and specificity of 86% and 78% (AUC 0.88-0.92, p < 0.0001). Inter and intra-observer variability for VTI measurements was excellent (intraclass correlation coefficients > 0.95). MRI and other echo measurements of LV mass and function as well as PA pressure gradient did not correlate with outcome.
Conclusion: PA VTI is a readily available and highly reproducible measurement that reflects morphologic LV performance and predicts outcomes in CCTGA patients better than conventional MRI assessment.
Article Information
vol. 130 no. Suppl 2 A19791
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Shreya Moodley;
- Sowmya Balasubramanian;
- Theresa Tacy;
- Frank Hanley;
- Frandics Chan;
- Rajesh Punn
- Pediatric Cardiology, Lucile Packard Children’s Hosp, Stanford, Palo Alto, CA
Abstract 19471: Holodiastolic Flow Reversal at the Descending Aorta is Neither Sensitive Nor Specific for Significant Aortic Regurgitation: a Phase Contrast Magnetic Resonance Study
Matthew A Harris, Mark A Fogel, Daniel W Kim, Timothy S Kim, Julian D Rose, Marc S Keller, Gregory L Fu, Kevin K Whitehead
Circulation. 2014;130:A19471
Abstract
Introduction: Holodiastolic flow reversal (HDR) measured at the descending aorta (DAO) has been used as a surrogate marker to identify significant aortic regurgitation using echocardiography.
Hypothesis: The purpose of this study is to determine if the presence of HDR correlates with the aortic valve regurgitant fraction (AVRF).
Methods: We retrospectively reviewed 167 CMR studies (64% male, 36% female) from January 2011 to May 2012 where velocity mapping was acquired at both the aortic valve and the DAO at the level of the diaphragm. Patients with coarctation of the aorta or single ventricle physiology were excluded from the study. Descending aortic velocity maps were checked for baseline offset using a static reference region. HDR was defined as flow reversal throughout diastole both before and after baseline correction. Significant aortic regurgitation was defined as an AVRF >10%.
Results: There were 145 patients (mean patient age was 14.1 ± 9.5 yrs) with an AVRF 20% (Group C) (Figure 1). Though the AVRF was significantly higher for HDR versus non-HDR pts (8.5 ± 14.2 vs 3.8 ± 6.6%, p=0.04), HDR was present in 32 Group A pts (22%). In comparison, 4 Group B pts (57%), and 7 Group C pts (47%) had HDR. Of the 64 Group A pts with either Tetralogy of Fallot (48) or Transposition (16), 15 pts (23%) had HDR. The sensitivity of HDR to predict the combined Groups B & C
was 0.5, and the specificity for Group A was 0.78.
Conclusions: DAO HDR is neither a very sensitive nor specific finding for predicting significant aortic regurgitation. HDR in the absence of significant aortic regurgitation appears to be a relatively common finding, especially in patients with repaired conotruncal anomalies. HDR should be interpreted with caution when evaluating aortic insufficiency, and likely has no role in the pediatric and young adult population or in patients after repair of conotruncal anomalies.
Article Information
vol. 130 no. Suppl 2 A19471
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Matthew A Harris;
- Mark A Fogel;
- Daniel W Kim;
- Timothy S Kim;
- Julian D Rose;
- Marc S Keller;
- Gregory L Fu;
- Kevin K Whitehead
- Pediatrics and Radiology, Childrens Hosp Philadelphia, Philadelphia, PA
Abstract 18799: Correlation of 2D Strain for Intraoperative Assessment of Right Ventricular Mechanics with Admittance Catheters in Juvenile Pigs
Sandhya R Ramlogan, Richard A Hopkins, Stephen F Kaine, Eric Buse, Suma P Goudar, Hongying Dai, Girish S Shirali
Circulation. 2014;130:A18799
Abstract
Background: Surgical manipulations such as opening the chest and volume loading have the potential to alter ventricular function. There is paucity of data on the effects of such manipulations on right ventricular (RV) function. We measured tricuspid annular plane excursion (TAPSE) and RV strain by two-dimensional echo (2DE) speckle tracking to assess systolic and diastolic RV function in juvenile pigs under conditions that simulate the intraoperative environment. These were compared to simultaneously obtained parameters of RV function using admittance catheters.
Methods: Epicardial 2DE was performed (Philips iE33) in 6 juvenile pigs (19-23kg). TAPSE and systolic and diastolic 2D RV longitudinal strain (LS) and strain rate (LSR) (TomTec Image Arena, TomTec, Germany) were measured offline. RV systolic and diastolic pressure/volume data was acquired simultaneously using admittance catheters (Scisense Inc, Canada) under conditions including closed chest, open chest and volume loading. Comparisons were made using Pearson’s correlation coefficient; heart rate and end-diastolic pressure (EDp) were stratified into tertiles to evaluate for threshold effects.
Results: We obtained 15 sets of paired data (Table). There was a high correlation between RVLS and EDp; this increased at lower EDp, at higher heart rates and with chest open vs. closed. There was high correlation between RVLS and TAPSE; this correlation was higher at higher heart rates, with the chest open and after a fluid bolus. Systolic LSR, diastolic SR and diastolic LSR did not correlate with catheter-derived measures of RV function.
Conclusions: 2D RV strain correlates well with RVEDp in conditions that simulate the intraoperative environment. We speculate that the increased correlations after sternotomy may be related to alterations in RV compliance, possibly related to the removal of pericardial restraint. The effect of altered loading conditions on echo-cath correlations bears further study.
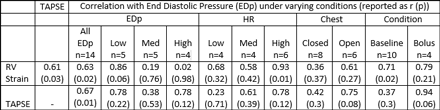
Article Information
vol. 130 no. Suppl 2 A18799
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Sandhya R Ramlogan;
- Richard A Hopkins;
- Stephen F Kaine;
- Eric Buse;
- Suma P Goudar;
- Hongying Dai;
- Girish S Shirali
- Heart Cntr, Children’s Mercy Hosp, Kansas City, MO
Abstract 18302: Longitudinal Evaluation of the Diastolic to Systolic Time-velocity Integral Ratio as a Doppler Derived Measure of Pulmonary Regurgitant Fraction in Patients With Surgically Repaired Tetralogy of Fallot
Misha Bhat, Elizabeth Goldmuntz, Laura Mercer-Rosa
Circulation. 2014;130:A18302
Abstract
Background: [[Unable to Display Character: –]] Pulmonary regurgitation (PR) following surgical repair for tetralogy of Fallot (TOF) is a major determinant of outcome but can be difficult to quantify by echocardiogram. We previously demonstrated a moderate correlation between the pulmonary artery diastolic-systolic time-velocity integral ratio (DSTVI) measured by echocardiogram and regurgitant fraction (RF) measured on cardiac magnetic resonance (CMR). We sought to investigate the ability of the DSTVI to identify temporal changes in PR.
Methods and Results: We conducted a longitudinal study of patients with baseline and follow up echocardiogram and CMR. Baseline studies were performed within 3 months of each other as part of a cross sectional research protocol. Follow up studies were performed <=7 months apart in cases with no interim interventions for PR. The DSTVI was calculated using Doppler interrogation in the main pulmonary artery. Wilcoxon matched-pairs signed-rank test was used to test whether changes in baseline to follow up DSTVI corresponded to changes in baseline to follow up PR. Linear regression of RF was fit on DSTVI to identify corresponding values of DSTVI.
Thirty-five subjects were included, age 18.3±3.5 years at follow up, 88.9% males. The time from baseline to follow up CMR was 60 months (interquartile range 46-73). On follow up, the DSTVI was 0.80 (±0.46) and RF was 35.5% (±17.8) with a moderate correlation between DSTVI and PR measured as RF by CMR (R=0.62, p=0.0001). A CMR RF of 20% and 40% (cut offs for moderate and severe PR) corresponded with a DSTVI of 0.52 (95% CI: 0.39[[Unable to Display Character: –]]0.66) and 0.79 (95% CI: 0.69[[Unable to Display Character: –]]0.89), respectively. Using the median DSTVI of 0.68 as a cutoff, DSTVI of <= 0.68 corresponded to a RF of 28% (20-37), whereas DSTVI > 0.68 corresponded to a RF of 44% (38-49), p=0.0001
Compared to the baseline study, there was no overall change in either DSTVI (p=0.61) or PR (p=0.89) at follow up and similarly, there was no difference in individual changes of RF and DSTVI (p=0.75).
Conclusions: [[Unable to Display Character: –]] The DSTVI provides an alternative and consistent quantitative measure of PR to RF by CMR that could be incorporated into the routine assessment of PR by echocardiogram. In addition, this index may help identify patients with TOF in need of early screening by CMR.
Article Information
vol. 130 no. Suppl 2 A18302
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1The Cardiac Cntr, The Children’s Hosp of Philadelphia, Philadelphia, PA
- 2Pediatrics, The Children’s Hosp of Philadelphia, Philadelphia, PA
Abstract 16887: Impact of Variability in Echocardiographic Interpretation on Assessment of Repair Following Congenital Heart Surgery
Anitha Parthiban, Jami C Levine, Meena Nathan, Jennifer A Marshall, Girish S Shirali, Steve D Simon, Steven D Colan, Jane W Newburger, Geetha Raghuveer
Circulation. 2014;130:A16887
Abstract
Background: Technical Performance Score (TPS), a tool based largely on the presence and magnitude of residua on postoperative echocardiograms (echo), has been used for assessing surgical repair and correlates with outcomes. The reproducibility of the echo measures that drive TPS classification has not been tested. We evaluated reader variability for echo components of TPS for tetralogy of Fallot (TOF) repair and arterial switch operation (ASO) in 2 centers and measured its effect on TPS.
Hypothesis: Inter-reader echo measurement variability will not substantially impact TPS classification.
Methods: Postoperative echos were evaluated in 67 subjects (39 TOF and 28 ASO). Two readers (1 per center) read each echo, blinded to center of origin. To assess intra-reader variability, 25% of echoes were re-reviewed by each reader. Measurements between readers were compared with Intra-class correlation (ICC). TPS Class (1 Optimal no residua, 2 Adequate minor residua, 3 Inadequate major residua) was assigned for each echo review by an independent investigator. The impact of measurement variability on overall TPS variability was compared using weighted Kappa (K) and % raw agreement.
Results: ICC was highest for Doppler velocity data and lower for measurements of small linear structures such as septal defects and vena contracta Figure. Overall TPS demonstrated good agreement (between reader TOF K = 0.82 and ASO K = 0.81). The 2 readers were concordant for TPS Class for 53 subjects (79%) and discordant for Classes 2 vs. 3 in 6 (9%); no readings were discordant between Classes 1 and 3 Table.
Conclusions: Although overall TPS demonstrated good agreement, inter-reader variation for echo measurements had a small, but important effect on TPS for ASO and TOF, particularly for the distinction between minor and major residua. Future studies of generalizability and reproducibility of TPS across centers and lesions are needed before TPS could be adopted as a national quality measure.
Article Information
vol. 130 no. Suppl 2 A16887
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Anitha Parthiban1;
- Jami C Levine2;
- Meena Nathan3;
- Jennifer A Marshall4;
- Girish S Shirali1;
- Steve D Simon5;
- Steven D Colan6;
- Jane W Newburger2;
- Geetha Raghuveer1
- 1Cardiology, Children’s Mercy Hosp and Clinics, Kansas City, MO
- 2Cardiology, Boston Children’s Hosp, Boston, MA
- 3Cardiac Surgery, Boston Children’s Hosp, Boston, MA
- 4Heart Cntr, Children’s Mercy Hosp and Clinics, Kansas City, MO
- 5Biostatistics, Univ of Missouri Kansas City Sch of Medicine, Kansas City, MO
- 6Cardiology, Boston Children’s Hosp, Boston, MO
Abstract 16964: Triplane Measurement of Fractional Area Change to Assess Single Right Ventricular Function
Muhammad Y Qureshi, Chelsea L Reece, Angela R Miller, Rebecca K Lindquist, Patrick W O’Leary
Circulation. 2014;130:A16964
Abstract
Background: Echocardiographic (echo) functional assessment of single systemic right ventricle (RV) lacks a reliable parameter and does not provide a substitute of volumetric ejection fraction (EF). RV volumes by 3D-echo are challenging and not widely used. Apical RV fractional area change (FAC) has shown some relation to CMR EF, but the strength of correlation has been suboptimal. Adding additional planes to apical FAC may improve this correlation. Our objective was to assess correlation of triplane FAC (apical, short-axis, and inflow-outflow) with CMR EF and to compare the tri- and single plane methods of measuring FAC to each other.
Methods: Subjects with hypoplastic left heart syndrome (after superior or total cavopulmonary anastomosis) were prospectively recruited. CMR was performed and right ventricular ejection fraction was calculated. Transthoracic echo studies were performed close to the time of the CMR scan (median interval: 1 d). FAC was measured in apical 4-chamber view, parasternal short axis view at the mid ventricular level, and para-apical right ventricular inflow-outflow view. Triplane FAC was calculated by the average of the three FAC. Comparison was made between FAC and CMR-derived ejection fraction.
Results: A total of 25 subjects underwent testing. Triplane FAC could not be assessed in 5, due to lack of optimal acoustic windows. Mean age was 10 ± 8 y (range 9 m to 24 y). Out of the uniplanar methods, apical FAC had the closest relationship to CMR EF. Triplane FAC showed even better correlation coefficient and R2 values; although in this small group the difference did not reach significance. Results are summarized in the Table.
Conclusion: In patients with single systemic RV, triplane FAC offers improved correlation with CMR EF relative to single plane evaluations. This approach may be useful if 3D echo is unavailable or of suboptimal quality and warrants further study.
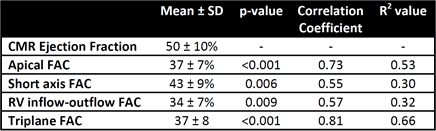
Article Information
vol. 130 no. Suppl 2 A16964
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Pediatric Cardiology, Mayo Clinic, Rochester, MN
- 2Div of Cardiovascular Diseases, Mayo Clinic, Rochester, MN
Abstract 15746: Statistical Shape Modeling of the Right Ventricle Differentiates Stages of RV Remodeling in Children With Pulmonary Hypertension: An Initial Analysis on 26 Patients
Frank Agyei-Ntim, Kendall Hunter, Uyen Truong, D D Ivy, Robin Shandas
Circulation. 2014;130:A15746
Abstract
Introduction: Current assessments of right ventricle (RV) dysfunction in pediatric pulmonary hypertension (PH) are limited by the lack of correlative data between RV shape changes and hemodynamic markers. Given the complexity of directly analyzing complete 4D RV image information, statistical shape approaches hold particular promise for defining principal shape features in large patient datasets. The first aim of this study was to identify the principal deformation features or modes of the RV in a small cohort of pediatric patients with varying degrees of PH. A subsequent aim was to evaluate correlations between principal RV deformation modes and hemodynamic markers.
Methods and Results: We selected 26 patients (aged 4 months – 19 years) who had undergone cardiovascular magnetic resonance imaging and right heart catheterization. Indexed pulmonary vascular stiffness (PVSi), indexed pulmonary vascular resistance (PVRi), cardiac index (CI) and ventricular-vascular coupling ratio (VVCR) were measured. Statistical shape analysis was used with principal component analysis (PCA) and a linear regression model to correlate principal RV deformation modes with PVSi and PVRi. The statistical shape analysis revealed four dominant deformation modes. Modes 1 and 2 captured RV basal dilatation and altered RV septal geometry respectively whilst modes 3 and 4 encoded RV apical rounding and wall thickening. Mode 1 and Mode 2 were associated with increasing PVRi: R = 0.42, R=0.51 respectively whilst Mode 3 and Mode 4 correlated to increasing PVSi: R=0.39, R=0.5. Mode 2 and mode 4 negatively correlated with increasing VVCR (R=-0.59, R=-0.47 respectively) whilst decreasing CI (R=-0.39) showed a statistical correlation only to mode 4.
Conclusions: Decreasing RV compliance (PVSi) is more associated with wall thickening (mode 4) than septal motion (mode 2), whilst increasing RV resistance (PVRi) appears more closely related to septal motion (mode 2) than wall thickening (mode 4). Modes 1, 3 and modes 2, 4 could separate different levels of RV remodeling (mild-to-moderate RV remodeling for modes 1, 3; moderate-to-severe RV remodeling for modes 2, 4). This approach may allow differentiation amongst pediatric PH patients undergoing adaptive versus maladaptive RV modeling.
Article Information
vol. 130 no. Suppl 2 A15746
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Dept of Bioengineering, Univ of Colorado Anschutz Med Campus, Aurora, CO
- 2Heart Institute, Children’s Hosp Colorado, Aurora, CO
- 3Dept of Pediatrics, Children’s Hosp Colorado, Aurora, CO
Abstract 15785: The Natural History of Doppler-Derived Left Ventricular Outflow Tract Gradients in Patients With Congenital Valvar Aortic Stenosis Before and After Balloon Valvuloplasty
Joseph D Kuebler, Jill Shivapour, Kimberlee Gauvreau, Steven D Colan, Doff B McElhinney, David W Brown
Circulation. 2014;130:A15785
Abstract
Introduction: Congenital aortic stenosis (AS) has been reported to manifest a slow rate of progression in mild disease with a greater likelihood of progression in patients with moderate-severe disease. The natural history of the Doppler estimated peak gradient (DEPG) in patients after balloon aortic valvuloplasty (BAV) has not previously been studied on a large scale.
Methods: A retrospective review was performed of 360 patients from 1984-2012 with AS providing a total of 2051 echocardiograms before and after BAV. Patients were excluded if they had an intervention within the first 30 days of life. The relationships between the AS DEPG and several predictors (age at time of initial echocardiogram, valve morphology, and history of intervention) were explored using linear mixed effect models. The DEPG slope was then calculated in patients who had at least 2 echocardiograms before and after balloon dilation using linear regression modeling.
Results: The rate of increase in the DEPG for all patients with AS was 5.6 mmHg per 10 years of age (p<0.001). The DEPG increased over time regardless of age at presentation with the greatest mean increase in patients presenting from 10-14.9 years (n=59; 11.9 mmHg per 10 years; p<0.001). Patients who went on to have a BAV or surgical intervention on the aortic valve had a significantly higher rate of AS progression than the overall patient cohort (n=59; 18.0 mmHg/10 years and n=36; 13.1 mmHg/10 years). Patients with a unicommissural (n=39) aortic valve had a significantly higher rate of progression compared to those with a bicommissural (n=270) aortic valve (8.1 mmHg/10 years and 4.5 mm Hg/10 years; p<0.001). The median rate of progression in the post-BAV group was significantly lower than the median pre-BAV rate of progression (n=34; pre-BAV 3.97 (1.69-8.7) mmHg/year; post-BAV 0.40 (-1.80-3.88) mmHg/year; p<0.01).
Conclusions: The DEPG of native valve congenital aortic stenosis shows a slow, linear rate of progression prior to intervention. The rate of progression is significantly higher in patients with a unicommissural aortic valve as well as those patients that go on to have a BAV and/or surgical intervention. The rate of the DEPG progression is significantly lower after BAV.
Article Information
vol. 130 no. Suppl 2 A15785
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Joseph D Kuebler1;
- Jill Shivapour2;
- Kimberlee Gauvreau1;
- Steven D Colan1;
- Doff B McElhinney3;
- David W Brown1
- 1Cardiology, Boston Children’s Hosp, Boston, MA
- 2Pediatric Cardiology, UH Rainbow Babies and Children’s Hosp, Cleveland, OH
- 3Pediatrics, Medicine, and Cardiothoracic Surgery, NYU Langone Med Cntr, New York, NY
Abstract 15519: Fluid Dynamics and Flow Profiles in the Great Arteries in Patients with Transposition of the Great Arteries (TGA) after Arterial Switch Operation with or without Lecompte Maneuver on Long-term Follow-up
Carsten Rickers, Kheradvar Arash, Ahmad Falahatpisheh, Philip Wegner, Dominik Gabbert, Christopher Hart, Inga Voges, Hans Heiner Kramer, Hans Hinerk Sievers
Circulation. 2014;130:A15519
Abstract
Background: Complex fluid dynamics (vorticity, helicity and wall shear stress) in the transposed great arteries in TGA patients after arterial switch operation (ASO) with spiral course or anterior branching of the pulmonary arteries (Lecompte) has been scarcely studied.
The aim of this study was a comprehensive assessment of blood flow in the great arteries utilizing advanced MRI techniques two decades after ASO.
Methods: 15 TGA patients (Lecompte: n=9, 20.7±2.3 yrs post ASO; non-Lecompte, spiral: n=6, 21.0±1.5 yrs post ASO; single surgeon) and 5 healthy volunteers (22.9±2.2) were studied with high field MRI at 3 Tesla. Blood flow dynamics were calculated from time-resolved 3D flow measurements (“4D flow”) using novel phase contrast MR-techniques (FOV 250-337 mm2, venc 150 cm/s in 3 othogonal directions, true spatial resolution: 2.5mm3isotropic, temp. resol 35 ms, TR/TE 4.6/3.2; α=5-10°). A dedicated software was used for colour coded 4D visualisation and grading of blood flow profiles and streamlines (GT-Flow™, Gyrotools Inc., Zurich). Vorticity, helicity and wall shear stress were calculated with our own custom-developed software. Additionally, a comprehensive anatomical and functional cardiovascular MR-protocol was applied to all patients and controls.
Results: In patients with a spiral course (non-Lecompte) of the great arteries, vorticity index and helicity were more favourable as compared to the Lecompte group (Aorta: 234.51±35 vs. 289.36±24 m2/s, pulmonary artery: 72.54±15 vs. 93.53±13 m2/s; p<0.01). With increased cross sectional area of the ascending aorta, vorticity was increased and shear stress correlated inversely (p<0.05). Flow measurements showed a significantly reduced cross-sectional area of the left pulmonary artery with impaired blood flow after Lecompte (p<0.01).
Conclusions: Two decades after operation, a spiral course of the great arteries in TGA patients post ASO showed more physiologically normal blood flow dynamics and balanced pulmonary flow as compared to those with anterior branching of the pulmonary arteries (post Lecompte). Therefore, in elegible patients, a spiral course should be considered before ASO.
Article Information
vol. 130 no. Suppl 2 A15519
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Carsten Rickers1;
- Kheradvar Arash2;
- Ahmad Falahatpisheh3;
- Philip Wegner1;
- Dominik Gabbert1;
- Christopher Hart1;
- Inga Voges1;
- Hans Heiner Kramer1;
- Hans Hinerk Sievers4
- 1Congenital Heart Disease and Pediatric Cardiology, Univ Hosp Kiel, Kiel, Germany
- 23Edwards Lifesciences Cntr for Advanced Cardiovascular Technology, UC-Irvine, Irvine, CA
- 3Edwards Lifesience Cntr, Univ California Irvine, Kiel, Germany
- 4Cardiac Surgery, Univ Hosp Lübeck, Lübeck, Germany
Abstract 19801: Creation of a Risk Adjustment Model for Adverse Events after Congenital Cardiac Catheterization
Natalie M Jayaram, Robert H Beekman, Lee N Benson, Ralf J Holzer, Kathy J Jenkins, Yan Li, Gerard R Martin, John W Moore, Richard Ringel, Jonathan J Rome, John A Spertus, Robert N Vincent, Lisa Bergersen
Circulation. 2014;130:A19801
Abstract
Background: As US healthcare increasingly focuses upon outcomes to quantify quality, there is a growing demand for risk models that can account for patient variability at different hospitals so that equitable comparison between institutions can be made. We developed a method for risk-standardizing adverse outcomes following congenital cardiac catheterization.
Methods: Using the national, multicenter, IMPACT® (Improving Pediatric and Adult Congenital Treatment) Registry, all patients undergoing diagnostic or interventional cardiac catheterization between January 2011 and March 2013 were identified. Multivariable hierarchical logistic regression was used to identify patient and procedural characteristics predictive of experiencing a major adverse event (see Figure) following cardiac catheterization.
Results: A total of 19,608 cardiac catheterizations were performed between January 2011 and March 2013. Among all cases, a major adverse event occurred in 378 (1.9%). After multivariable adjustment, 8 variables were identified as critical for risk-standardization: patient age, renal insufficiency, single-ventricle physiology, procedure-type risk group, low systemic saturation, low mixed venous saturation, elevated systemic ventricular end diastolic pressure, and elevated main pulmonary artery pressures (Figure). The model had good discrimination (C-statistic of 0.70), confirmed by bootstrap validation (validation C-statistic of 0.69) and had excellent calibration (slope= 0.97; standard error 0.041; p-value [for difference from 1]= 0.42).
Conclusion: In a large national registry, we developed and validated a model to predict major adverse events following diagnostic or interventional cardiac catheterization for congenital heart disease. This model can support more equitable comparisons between hospitals and can facilitate national quality improvement efforts.
Article Information
vol. 130 no. Suppl 2 A19801
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Natalie M Jayaram1;
- Robert H Beekman2;
- Lee N Benson3;
- Ralf J Holzer4;
- Kathy J Jenkins5;
- Yan Li6;
- Gerard R Martin7;
- John W Moore8;
- Richard Ringel9;
- Jonathan J Rome10;
- John A Spertus6;
- Robert N Vincent11;
- Lisa Bergersen5
- 1Cardiovascular Outcomes Rsch, Saint Luke’s Mid America Heart Institute and Children’s Mercy Hosps and Clinics, Kansas City, MO
- 2Div of Cardiology, Cincinnati Children’s Hosp Med Cntr, Cincinnati, OH
- 3Div of Cardiology, The Hosp for Sick Children, Toronto, Canada
- 4Div of Cardiology, Nationwide Children’s Hosp, Columbus, OH
- 5Div of Cardiology, Boston Children’s Hosp, Boston, MA
- 6Cardiovascular Outcomes Rsch, Saint Luke’s Mid America Heart Institute, Kansas City, MO
- 7Div of Cardiology, Children’s National Health System, Washington D.C., DC
- 8Div of Cardiology, Rady Children’s Hosp and UCSD Sch of Medicine, San Diego, CA
- 9Div of Cardiology, Johns Hopkins Children’s Cntr, Baltimore, MD
- 10Div of Cardiology, Children’s Hosp of Philadelphia, Philadelphia, PA
- 11Div of Cardiology, Children’s Healthcare of Atlanta and Emory Univ Sch of Medicine, Atlanta, GA
Abstract 19605: Aortic Valve Morphology Influences Outcomes of Balloon Aortic Valvuloplasty in Children with Congenital Aortic Stenosis
Christopher J Petit, Kevin Gao, Bryan H Goldstein, Sean M Lang, Sung I Kim, Ritu Sachdeva
Circulation. 2014;130:A19605
Abstract
Background: In isolated congenital aortic valve stenosis (AS) early and late outcomes following BAV vary widely despite near uniform procedural and technical factors. Anatomic factors may account for some of this variability in outcomes. We sought to identify morphologic factors that may influence outcomes of BAV.
Hypothesis: Aortic valve anatomy influences outcomes after BAV.
Methods: We reviewed all pts who underwent BAV at two large pediatric institutions between 2007-2013. Pre- and post-BAV echocardiography (echo) was reviewed. Anatomic indices including valve leaflet excursion, valve annulus area (AA), valve orifice area (OA), valve morphology, and raphe (fusion) length were measured on all pre- and post-BAV echo. Primary endpoint was favorable tear (FT), defined as tear along a commissure. Unfavorable tear (UT) was defined as tear into a leaflet, and no tear (NT) defined as no visible tear. Secondary endpoint was aortic insufficiency increase ≥2 degrees (AI+).
Results: A total of 88 pts had complete clinical and echo data available, of whom 34 (39%) were neonates at the time of BAV. FT was noted in 56 (58%) pts, UT noted in 16 pts (18%), and NT in 16 pts (18%). FT was more common in functionally unicuspid valves (p=0.02), with lower OA/AA ratio (p=0.003), and in pts with greater fusion length (p=0.042). OA was highly correlated with fusion length (p<0.001) and aortic root diameter (p<0.001) but not with leaflet excursion (p=0.68). Of 16 pts with AI+, 11 were in the UT cohort (11/16), while 5 were in the FT cohort (5/56) (OR for AI+ 13.7, 3.8-50). Pts with larger OA (p=0.007) and less fusion (p=0.032) were more likely to develop UT. The cath gradient fell from a median of 57mmHg (IQR 48-75) to 23mmHg (IQR 16-31) after BAV ; gradient reduction was lower in UT (23.2, 12-33) than in FT (30.9, 24-37) or UT (39.0, 27-51) (p=0.042). Multivariate analysis demonstrated that functionally unicuspid valve morphology (p=0.048) and lower OA (p=0.030) were associated with FT.
Conclusion: Favorable and unfavorable tears post-BAV are associated with similar gradient relief but discrepant degrees of AI. FT and UT may be predictable based upon valve morphology. Enhanced BAV patient selection with careful assessment of anatomic valve metrics may result in improved outcomes following BAV.
Article Information
vol. 130 no. Suppl 2 A19605
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Pediatrics, Emory Univ Sch of Medicine, Atlanta, GA
- 2Pediatrics, Univ of Cincinnati Sch of Medicine, Cincinnati, OH
- 3Pediatrics, Univ of Cincinnati College of Medicine, Cincinnati, OH
Abstract 18126: Calcium Channel Blockers Accelerate Aortic Aneurysm and Cause Premature Lethality in Marfan Syndrome and Related Conditions
Jefferson J Doyle, Alexander J Doyle, Nicole K Wilson, Jennifer P Habashi, Djahida Bedja, Ryan E Whitworth, Mark E Lindsay, Loretha Myers, Nick Huso, Suha Bachir, David Huso, Daniel P Judge, Harry C Dietz
Circulation. 2014;130:A18126
Abstract
Calcium channel blockers (CCBs) are prescribed to patients with Marfan syndrome and related inherited thoracic aortic aneurysm (TAA) syndromes for prophylaxis against TAA progression, despite limited evidence for their efficacy in these disorders. Unexpectedly, Marfan mice treated with CCBs showed accelerated TAA expansion, rupture and premature lethality. In collaboration with other members of the GenTAC consortium, we conducted a case-control study to assess the effect of CCBs in humans with Marfan syndrome and related TAA conditions. These primarily included Loeys-Dietz, Turner and Ehlers-Danlos syndromes, familial thoracic aortic aneurysm, and bicuspid aortic valve with aneurysm. Marfan patients with native aortic roots at the time of enrollment who received CCBs (compared to other antihypertensive agents) had an increased risk of aortic dissection (odds ratio (OR) 12.5, p<0.05). Strong trends were maintained after correction for blood pressure (OR 12.7, p=0.06) or aortic root size (OR 11.2, p=0.08) at enrollment. A more profound effect of CCBs on outcome could be masked by the practice of intervening with aortic surgery when the aorta reaches a dimension that confers risk for dissection. In keeping with this hypothesis, we found that CCB-treated Marfan patients had an increased risk of aortic surgery (OR 5.5, p<0.001) when compared to patients on other antihypertensive agents, which remained significant when corrected for either blood pressure (OR 5.4, p<0.001) or aortic size (OR 5.0, p<0.01) at enrollment. For patients with other forms of TAA and native aortic roots at the time of enrollment (n=1,819), there was suggestion of an increased risk for aortic dissection in those who had taken CCBs, although this did not reach statistical significance (OR 4.7, p=0.26). This was most likely secondary to prophylactic surgical intervention, given that these patients again had an increased risk of aortic surgery (OR 2.4, p=0.004), which remained significant when corrected for either blood pressure (OR 2.2, p=0.016) or aortic size (OR 2.2, p=0.017) at enrollment. These data indicate that CCB treatment exacerbates aortic disease in Marfan syndrome and related TAA conditions, and suggests that CCBs should be used with caution in these patient populations.
Article Information
vol. 130 no. Suppl 2 A18126
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Jefferson J Doyle1;
- Alexander J Doyle1;
- Nicole K Wilson1;
- Jennifer P Habashi1;
- Djahida Bedja1;
- Ryan E Whitworth2;
- Mark E Lindsay3;
- Loretha Myers1;
- Nick Huso1;
- Suha Bachir1;
- David Huso1;
- Daniel P Judge1,
- GenTAC Consortium members;
- Harry C Dietz1
- 1Institute of Genetic Medicine, Johns Hopkins Univ, Baltimore, MD
- 2RTI International, RTI International, Durham, NC
- 3Medicine and Pediatrics, Massachusetts General Hosp Thoracic Aortic Cntr, Boston, MA
Abstract 17042: Surgery and Outcomes for Transposition of the Great Arteries among Developing World Congenital Heart Surgery Programs: a Report from the International Quality Improvement Collaborative
David N Schidlow, Kathy J Jenkins, Kimberlee Gauvreau, Ulisses A Croti, Do Thi Cam Giang, Nestor F Sandoval, Rama K Konda, Aldo Castaneda
Circulation. 2014;130:A17042
Abstract
Objectives: Surgical care for CHD in the developing world is rapidly evolving, allowing an opportunity for survival for children with previously lethal conditions. Little information is available about such patients and their outcomes. The objectives of this study were to identify (1) patient characteristics, (2) surgical interventions, (3) institutional characteristics, and (4) risk factors for mortality among patients in the International Quality Improvement Collaborative (IQIC) undergoing surgery for transposition of the great arteries (TGA) with intact ventricular septum (IVS) and TGA with ventricular septal defect (VSD).
Methods: We utilized a novel international collaborative database collected by developing world congenital heart surgical programs as part of a QI project. All TGA (IVS and VSD) cases from 2010-2012 were included. Demographic, surgical and institutional characteristics and their associations with in-hospital mortality were identified.
Results: There were 559 patients at 21 centers who underwent surgery for TGA: 348 TGA/IVS and 211 TGA/VSD. TGA/IVS patients underwent arterial switch operation (ASO), 282 (81%); 2-stage ASO, 24 (7%); and atrial switch (ATS) 42, (12%). TGA/VSD patients underwent ASO, 169 (80%); 2-stage ASO, 19 (9%); and ATS, 23 (11%); all with VSD closure. In-hospital mortality ranged from 10% to 25% depending on procedure (Figure). Unadjusted mortality rates were higher among centers performing <10 TGA repairs yearly (OR 3.98; 95% CI 2.08-7.60, p<0.001). Among patients with TGA/IVS who underwent primary ASO, risk factors for mortality included male gender (p=0.05), weight <3 kg (p=0.04), and prematurity (p=0.007).
Conclusions: Infants with TGA in the developing world increasingly have access to complex surgical repairs, and the majority of such patients undergo single-stage ASO. Mortality remains substantial; however, multi-center collaborative QI efforts are likely to benefit these patients.
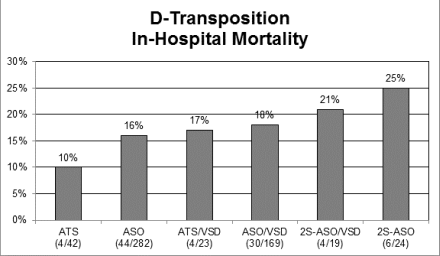
Article Information
vol. 130 no. Suppl 2 A17042
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- David N Schidlow1;
- Kathy J Jenkins1;
- Kimberlee Gauvreau1;
- Ulisses A Croti2;
- Do Thi Cam Giang3;
- Nestor F Sandoval4;
- Rama K Konda5;
- Aldo Castaneda6
- 1Cardiology, Boston Children’s Hosp, Boston, MA
- 2Surgery, Hosp da Criança e Maternidade de São José do Rio Preto, São José do Rio Preto, Brazil
- 3Pediatrics, Nhi Dong 1, Ho Chi Minh City, Viet Nam
- 4Pediatric Cardiac Surgery, Fundacion Cardioinfantil – IC, Bogota, Colombia
- 5Intensive Care, Care Hosp, Hyderabad, India
- 6Pediatrics, Fundacion Aldo Castaneda, Guatemala City, Guatemala
Abstract 16469: Outcomes After In-Utero Cardiac Interventions: A Preliminary Report of the Collaborative International Fetal Cardiac Intervention Registry
Anita J Moon-Grady, Michael Belfort, Ramen Chmait, Joanna Dangel, Roland Devlieger, Stephen Emery, Alberto Galindo, Ulrich Gembruch, Sofia Grinenco, Mounira Habli, Ulrike Herberg, Edgar Jaeggi, Mark Kilby, Pablo Marantz, Shaine Morris, Lucas Otaño, Carlos Pedra, Simone Pedra, Jay Pruetz, Ruben Quintero, Greg Ryan, Gurleen Sharland, John Simpson, Emanuel Vlastos, Wayne Tworetzky, Louise Wilkins-Haug, Dick Oepkes
Circulation. 2014;130:A16469
Abstract
Objective: Fetal cardiac intervention (FCI) for relief of semilunar valve stenosis or atrial level restriction has been reported in single-institution series promoting technical and physiologic success. No contemporary multi-center experience with FCI has been published. We describe creation of an international registry of cases presenting for possible FCI with the intention of compiling technical aspects and outcomes data.
Methods: The International Fetal Cardiac Intervention Registry (IFCIR) was established in 2010 to collect voluntarily submitted procedure-related data. For this initial descriptive analysis the database was queried for all entries, details of diagnosis, procedures performed, and outcomes. Maternal-fetal dyads referred since 2001 for possible FCI were included; a favorable outcome was defined as biventricular circulation for valvuloplasty and survival to discharge for atrial septal procedures.
Results: Of 372 cases included, 246 underwent FCI: 100 from a previous single-center report were not included in the present analysis. Among liveborn infants diagnosed in utero with evolving hypoplastic left heart syndrome, biventricular circulation was present more often when FCI was attempted (51% (95%CI 38-64%) v 23% (4-42%) p=0.02), but when procedure-related losses (n=15) and fetal demise (3) were counted as treatment failures in this cohort, the difference was no longer significant (biventricular in 36% of FCI group v 21% in non-FCI, p=0.16). Survival in fetuses with reported atrial restriction was similar with or without FCI (15/28 v 5/10), though non-uniform criteria for this diagnosis were noted in the analysis of the data.
Conclusion: In this initial report, we describe the content of the IFCIR, and present postnatal data that suggest a potential benefit to fetal therapy and support proposals for additional work in this area. Analyses pertaining to specific diseases and patient, center, and procedural variables are ongoing.
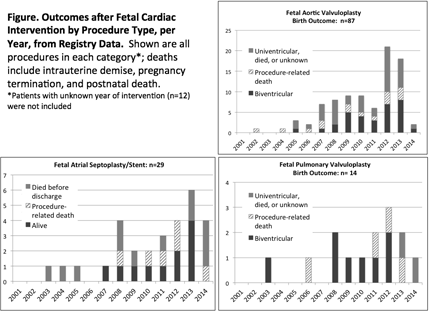
Article Information
vol. 130 no. Suppl 2 A16469
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Anita J Moon-Grady1;
- Michael Belfort2;
- Ramen Chmait3;
- Joanna Dangel4;
- Roland Devlieger5;
- Stephen Emery6;
- Alberto Galindo7;
- Ulrich Gembruch8;
- Sofia Grinenco9;
- Mounira Habli10;
- Ulrike Herberg11;
- Edgar Jaeggi12;
- Mark Kilby13;
- Pablo Marantz14;
- Shaine Morris15;
- Lucas Otaño16;
- Carlos Pedra17;
- Simone Pedra17;
- Jay Pruetz18;
- Ruben Quintero19;
- Greg Ryan20;
- Gurleen Sharland21;
- John Simpson22;
- Emanuel Vlastos23;
- Wayne Tworetzky24;
- Louise Wilkins-Haug25;
- Dick Oepkes26
- 1Pediatrics, Univ of California-San Francisco, San Francisco, CA
- 2Obstetrics and Gynecology, Baylor College of Medicine, Houston, TX
- 3Maternal-Fetal Medicine, Keck Sch of Medicine, Univ of Southern California, Los Angeles, CA
- 4Perinatal Cardiology, Perinatal Cardiology Clinic, Med Univ of Warsaw, Warsaw, Poland
- 5Obstetrics, Univ Hosps KULeuven, Leuven, Belgium
- 6Obstetrics and Gynecology, Magee Women’s Hosp of the Univ of Pittsburgh Med Cntr, Pittsburgh, PA
- 7Obstetrics and Gynecology, Hosp Universitaio 12 de Octubre, Madrid, Spain
- 8Obstetrics and Prenatal Medicine, Univ Clinics of Bonn, Bonn, Germany
- 9Pediatrics, Hosp Italiano de Buenos Aires, Buenos Aires, Argentina
- 10Pediatric Surgery, Cincinnati Children’s Hosp, Cincinnati, OH
- 11Pediatric Cardiology, Univ of Bonn, Bonn, Germany
- 12Pediatrics-Cardiology, Hosp for Sick Children, Toronto, Canada
- 13Obstetrics, Univ of Birmingham, Edgbaston, Birmingham, United Kingdom
- 14Cardiology, Hosp Italiano de Buenos Aires, Buenos Aires, Argentina
- 15Pediatrics, Baylor College of Medicine, Houston, TX
- 16Obstetrics, Hosp Italiano de Buenos Aires, Buenos Aires, Argentina
- 17Pediatrics-Cardiology, Hosp do Coração, São Paulo, Brazil
- 18Cardiology, Children’s Hosp of Los Angeles, Los Angeles, CA
- 19Obstetrics and Gynecology, Univ of Miami Miller Sch of Medicine, Miami, FL
- 20Obstetrics, Mount Sinai Hosp, Univ of Toronto, Toronto, Canada
- 21Fetal Cardiology, Evelina Children’s Hosp, London, United Kingdom
- 22Congenital Heart Disease, Evelina Children’s Hosp, London, United Kingdom
- 23Obstetrics and Gynecology, SSM Cardinal Glennon Children’s Med Cntr, St. Louis, MO
- 24Cardiology, Boston Children’s Hosp, Boston, MA
- 25Obstetrics and Gynecology, Brigham and Women’s Hosp, Boston, MA
- 26Obstetrics, Leiden Univ Med Cntr, Leiden, Netherlands
Abstract 16095: Is the Arachidonic Acid Pathway Adequately Inhibited in Shunted Infants on Aspirin?
Dongngan T Truong, Joyce T Johnson, David K Bailly, Jason R Clawson, Madolin K Witte, L. LuAnn Minich
Circulation. 2014;130:A16095
Abstract
Introduction: Prevention of shunt thrombosis is vital in infants who require a reliable source of pulmonary blood flow. Aspirin (ASA) prophylaxis is used to inhibit the platelet arachidonic acid (AA) pathway, but dosing is extrapolated from adults, and no therapeutic monitoring guidelines exist. We sought to determine the percentage of infants on ASA after shunt surgery with adequate AA inhibition using thromboelastography with platelet mapping (TEG-PM) and to describe adverse events (AE).
Methods: We performed a prospective observational study of infants treated with ASA after shunt (12/2012-5/2014). ASA was dosed at 3-5 mg/kg enterally starting postop day 1. TEG-PM was obtained at 5 intervals: 1) baseline (before cardiopulmonary bypass), 2) prior to 1st ASA dose, 3) after 3rd ASA dose, 4) 1st postop clinic visit, and 5) 2-6 months postop. Adequate AA inhibition was defined as ≥70%, the target parameter in the pediatric Berlin Heart protocol. Bleeding and thrombotic events were recorded as AEs. Clinicians were blinded to TEG-PM results. Fisher’s exact, t-test, and linear mixed model were used.
Results: The 25 subjects had cavopulmonary (N=11), aortopulmonary (N=9), and Sano (N=5) shunts; 9 (36%) were previously on ASA. Of the 20 who completed follow-up, 8 subjects had 13 AEs: 12 bleeding events (50% ≤7 days postop), 1 thrombotic stroke, and 0 shunt thrombosis. AA inhibition was ≥70% after the 3rd ASA dose in 10/21 (48%), at the 2 postop follow-ups in 4/15 (27%), and at all intervals on ASA in 1 (4%). ASA dose and time since dose were not associated with AA inhibition. Subjects with bleeding AEs had higher baseline ADP inhibition (76%±9 vs 44%±8, P=0.02), higher AA inhibition prior to 1st ASA dose (46%±22 vs 17%±15, P<0.01) and after the 3rd ASA dose (74%±20 vs 43%±33, P=0.04). TEG-PM nearest the bleeding AE (≤9 days) showed average ADP and AA inhibition of 76% and 51%.
Conclusions: AA inhibition was <70% in most shunted infants. No shunt thrombosis occurred but 1/3 had bleeding AEs, suggesting the target AA inhibition may be lower for the shunted infant. Increased ADP inhibition concomitant with AA inhibition predicted bleeding. Future studies will aim to determine the role of elevated ADP inhibition in bleeding risk and to determine the optimal AA target in this population.
Article Information
vol. 130 no. Suppl 2 A16095
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Dongngan T Truong1;
- Joyce T Johnson1;
- David K Bailly2;
- Jason R Clawson3;
- Madolin K Witte2;
- L. LuAnn Minich1
- 1Pediatric Cardiology, Univ of Utah, Salt Lake City, UT
- 2Pediatric Critical Care, Univ of Utah, Salt Lake City, UT
- 3Pediatrics, Univ of Utah, Salt Lake City, UT
Abstract 12910: Safety and Utility of Cardiac Catheterization in the Early Postoperative Period Following Congenital Cardiac Surgery
Kimberly I Mills, Lisa B Bergersen, Matthew Jolley, Sarah A Teele, John E Mayer, Joshua W Salvin
Circulation. 2014;130:A12910
Abstract
Introduction: In patients with clinical concern for residual anatomic or physiologic lesions after congenital cardiac surgery, the risk to benefit ratio of early postoperative cardiac catheterization (EPOC) remains unclear.
Methods: Retrospectively, patients undergoing EPOC (catheterization ≤42 days after surgery and prior to discharge) were identified from a single institutional database between 1/1/06 to 10/31/13. Patient, procedural and high-severity adverse event (HSAE) data were obtained from the Congenital Cardiac Catheterization Outcomes Project. Univariate analysis identified risk factors for HSAEs and a multivariable model was created to describe their relationship. Utility of EPOC was determined by re-intervention (surgical and/or catheter-based).
Results: In the cohort, 561 EPOCS occurred on median POD 10 (5, 19) in 413 subjects with median age of 4.6 mos (3.2, 9.9) and weight 4.4 kg (0.8, 18.2). The majority of cases were not elective (92%), performed on inotropes (62%) and mechanically ventilated (96%). Residual defects were found in 75% of the cases: 50% requiring catheter-based interventions, 15% necessitating re-operation and 10% underwent both catheter-based intervention and re-operation. There were 70 HSAEs (12.5%) including 1 death, 2 emergent surgeries and 5 ECMO cannulations. Vascular tears occurred at surgical anastomotic sites in 11 of 198 (6%) cases involving angioplasty. HSAEs were associated with low patient weight (p=0.02), interventional procedures (p=0.03), greater number of interventions (p<0.01), higher procedure risk group (p=0.03), blood transfusion (p<0.01) and longer case duration (p<0.01). In multivariable analysis, HSAEs were associated with case duration (OR 1.008, 95% CI 1.003-1.012), blood transfusion (OR 0.431, 95% CI 0.25-0.745) and male gender (OR 0.573, 95% CI 0.331-0.992). Time from surgery to EPOC was not associated with an increased rate of HSAEs (p=0.5).
Conclusions: EPOC carried an increased rate of HSAEs with no association between POD and the rate of HSAEs. EPOC identified significant residual defects in the majority of patients necessitating re-intervention. EPOC should be considered in patients with unexpected cardiopulmonary concerns following congenital cardiac surgery.
Article Information
vol. 130 no. Suppl 2 A12910
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Kimberly I Mills1;
- Lisa B Bergersen1;
- Matthew Jolley2;
- Sarah A Teele1;
- John E Mayer3;
- Joshua W Salvin1
- 1Dept of Cardiology, Boston Children’s Hosp, Boston, MA
- 2Dept of Anesthesia, Boston Children’s Hosp, Boston, MA
- 3Dept of Cardiac Surgery, Boston Children’s Hosp, Boston, MA
Abstract 12244: 48 Week Outcomes Using Targeted Biologic Inhibition Agents to Treat Aggressive Multivessel Pulmonary Vein Stenosis
Kara Western, Mark Kieran, Christopher Baird, Christina Ireland, Steven D Colan, Kimberlee Gauvreau, Audrey C Marshall, Laureen Sena, Kathy J Jenkins
Circulation. 2014;130:A12244
Abstract
Introduction: Intraluminal pulmonary vein stenosis (PVS) can occur in isolation, with congenital heart disease (CHD), chronic lung disease, or both. PVS can progress rapidly with vessel atresia, pulmonary hypertension, and RV failure, and can be fatal within months (mos) of diagnosis. Previous research has demonstrated that intraluminal obstruction occurs due to neointimal proliferation of myofibroblast-like cells, which express VEGF and PDGFR.
Hypothesis: Adjunct targeted inhibition of VEGF (bevacizumab) and PDGFR (imatinib mesylate) may reduce disease activity in patients (pts) with multivessel intraluminal PVS and prolong survival.
Methods: This is a single-arm, prospective, open-label FDA approved study using bevacizumab and imatinib mesylate to treat progressive multivessel intraluminal PVS. Pts with obstruction of ≥ 2 veins (echocardiographic (Echo) mean gradient ≥ 4 mmHg) were included after aggressive surgical or catheter based relief of obstruction. All but 3 pts had ≥ 1 PVS surgery prior to initiation. Pts with CHD received imatinib mesylate only, those without CHD or who progressed on study also received bevacizumab. Follow-up included lung scan and Echo q 4-8 weeks (wks); CT scan or angiography q 24 wks. We report initial (31 of 44 enrolled pts) who have reached 48 wks or study endpoint.
Results: Among 31 pts, 17 were treated for 48 wks; 3 transplanted; 11 discontinued drug < 48 wks. Mean age at diagnosis was 4 mos; drug initiation 9 mos. Two had isolated disease, 16 CHD, 3 lung disease, 10 both. Twenty-four pts received imatinib mesylate only; 7 both drugs. Median number of pulmonary veins involved was 4 (range 2-5); 12 pts had ≥ 1 atretic vessel at initiation. Pts received mean of 85% planned doses of drug. At 48 wks, 26/31 (84%) were alive, 2 post-transplant. Eight of 31 (26%) stabilized, 6/31 (19%) did not require any subsequent pulmonary vein intervention.
Conclusions: Multi-modal treatment including aggressive surgical and catheter based intervention and targeted inhibition of myofibroblast proliferation resulted in a 48 wk survival of 84% among pts with multivessel intraluminal PVS. These results suggest that targeted inhibition may play an important role in managing aggressive pulmonary vein disease.
Article Information
vol. 130 no. Suppl 2 A12244
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Kara Western1;
- Mark Kieran2;
- Christopher Baird1;
- Christina Ireland1;
- Steven D Colan1;
- Kimberlee Gauvreau1;
- Audrey C Marshall1;
- Laureen Sena1;
- Kathy J Jenkins1
- 1Cardiology, Boston Children’s Hosp, Boston, MA
- 2Pediatric Brain Tumors, Dana Farber Cancer Institute, Boston, MA
Abstract 12255: Up to Seven-Year Outcomes After Transcatheter Pulmonary Valve Replacement in the Prospective Multicenter US Melody Valve Investigational Device Exemption Trial
John P Cheatham, Julie A Vincent, Evan M Zahn, Lisa Bergersen, Darren P Berman, James E Lock, William E Hellenbrand, Thomas K Jones, Doff B McElhinney
Circulation. 2014;130:A12255
Abstract
Introduction: Transcatheter pulmonary valve replacement (TPVR) with the Melody valve was introduced in the US in 2007 through the Investigational Device Exemption (IDE) trial. Although earlier European and IDE reports showed good short-term outcomes, there are no published long-term follow-up (FU) data. The studies with the longest FU reported outcomes a median of ~2.5 yrs post-TPVR.
Methods and Results: The IDE trial enrolled 171 pts with postoperative right ventricular outflow tract (RVOT) conduit dysfunction from Jan 2007 to Jan 2010; the 148 pts (median age 19 yrs) who received a TPV and were discharged with the valve in place comprise the cohort for this study. During a median FU of 4.5 yrs (max 7 yrs), 4 pts died: 1 after endocarditis and RVOT reintervention, 3 unrelated to the TPV. A total of 32 pts underwent RVOT reintervention: initially TPV dilation in 7, redo TPVR in 18, and TPV explant in 7. Of 25 pts who underwent catheter-based reintervention, 8 had another intervention (transcatheter-4 or explant-4) a median of 1.5 yrs later. Reinterventions were for RVOT obstruction in 28 pts (with stent fracture in 25), endocarditis in 2 (1 with stenosis, 1 with regurgitation [PR]), and RV dysfunction in 2. In the 113 pts who were alive and reintervention-free (median FU 4.5 yrs), the most recent gradient was unchanged from early post-TPVR (17±7 vs 16±8mmHg, p=0.7), and all but 1 had mild or less PR (most trivial/none). Stent fractures were diagnosed in 50 pts, half of whom were reintervention-free and had good TPV function a median of 2 yrs later. At 5 yrs, freedom from RVOT reintervention and explant were 76±4% and 91±3%, respectively. A homograft conduit, no conduit pre-stent, discharge mean RVOT gradient >25mmHg, and pre-implant ≥moderate TR were associated with shorter freedom from reintervention on multivariable Cox regression. Almost all pts were in NYHA class I or II at FU.
Conclusions: TPVR with the Melody valve provided good hemodynamic and clinical outcomes 4-7 yrs after implant. Primary valve failure was rare. The main cause of TPV dysfunction was stenosis related to stent fracture, which was less common once pre-stenting became more widely adopted. Based on these findings, TPVR appears to be a good option for at least mid-term palliation of RVOT conduit dysfunction.
Article Information
vol. 130 no. Suppl 2 A12255
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- John P Cheatham1;
- Julie A Vincent2;
- Evan M Zahn3;
- Lisa Bergersen4;
- Darren P Berman5;
- James E Lock4;
- William E Hellenbrand6;
- Thomas K Jones7;
- Doff B McElhinney8
- 1Cardiology, Nationwide Children’s Hosp, Columbus, OH
- 2Pediatrics, Columbia Univ Med Cntr, New York, NY
- 3Pediatrics, Cedars-Sinai Heart Institute, Los Angeles, CA
- 4Cardiology, Children’s Hosp Boston, Boston, MA
- 5Cardiology, Miami Children’s Hosp, Miami, FL
- 6Pediatrics, Yale Sch of Medicine, New Haven, CT
- 7Pediatrics, Seattle Children’s Hosp, Seattle, WA
- 8Pediatrics, NYU Med Cntr, New York, NY
Abstract 20439: Novel Long Intergenic Noncoding RNAs Modulate Adipose Functions in Human
Xuan Zhang, Rachel Ballantyne, Chenyi Xue, Jane F Ferguson, Brian Gregory, Mingyao Li, Muredach P Reilly
Circulation. 2014;130:A20439
Abstract
Recently long intergenic noncoding RNAs (lincRNAs) have emerged as key mediators of cellular differentiation and functions in a variety of cell systems critical to cardiovascular and metabolic disorders. To identify and investigate novel functional lincRNAs in human adipose, we performed deep high-throughput RNAseq (>200 million reads/sample) in subcutaneous adiposes of 13 health volunteers. Of an integrated dataset of 54,944 human lincRNAs, 6,558 lincRNAs were detected.
Here we report 2 cytoplasmic adipose lincRNAs, linc-DMRT2 and linc-NFE2L3-1, were detected in human adipocytes but not monocytes or macrophages. Linc-DMRT2, one of the most abundant adipose lincRNAs, was markedly induced during in vitro human adipocyte differentiation. Notably, single molecule RNA FISH (fluorescence in situ hybridization) demonstrated that linc-DMRT2 were exclusively present in adipocyte cytoplasma and co-localized with processing bodies (P-bodies) marker, GW182, suggesting its potential role in modulating turnover of certain RNA species. In addition, linc-NFE2L3-1, predominantly detected in adipose and skeleton muscle, is localized near an established GWAS locus associated with waist-hip ratio adjusted BMI. We identified 4 SNPs in linc-NFE2L3-1 reaching genome wide significance for BMI (lead SNP rs10267498, P=2.73х10-8). Linkage disequilibrium analysis confirmed linc-NFE2L3-1 harbors stronger GWAS signals than protein-coding genes in the locus, suggesting lincRNA might be causal for GWAS association with BMI. Bioinformatic prediction algorithms identified potential binding sites in linc-DMRT2 and linc-NFE2L3-1 for multiple microRNAs that have been demonstrated to regulate adipogenesis (e.g. miR-15 a/b and let-7) or adipocyte functions (e.g. miR-320).
In summary, our data suggest that cytoplasmic linc-DMRT2 and linc-NFE2L3-1 may play important roles in adipocyte biology by functioning as competing endogenous RNAs and binding specific microRNAs that mediate adipocyte cellular functions. Genetic variation in such human linRNAs may contribute to cardiometabolic traits.
Article Information
vol. 130 no. Suppl 2 A20439
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Xuan Zhang1;
- Rachel Ballantyne1;
- Chenyi Xue1;
- Jane F Ferguson1;
- Brian Gregory2;
- Mingyao Li3;
- Muredach P Reilly1
- 1Cardiovascular Institute, Univ of Pennsylvania, Philadelphia, PA
- 2Dept of Biology, Univ of Pennsylvania, Philadelphia, PA
- 3Dept of Biostatistics and Epidemiology, Univ of Pennsylvania, Philadelphia, PA
Abstract 16656: DNA Methylation and mRNA Sequencing of Monocytes From a Large Cohort Identifies Associations Between an Epigenetic Biomarker of Smoking, AHRR Expression, and Atherosclerosis
Lindsay M Reynolds, Ma Wan, Jingzhong Ding, Jackson R Taylor, Kurt Lohman, R Graham Barr, Timothy D Howard, Dan Su, Devin Porter, Ryan Gimple, Gary S Pittman, David Siscovick, Bruce M Psaty, Steven Shea, David R Jacobs, Stephen S Rich, James E Hixson, James H Stein, Hendrik G Stunnenberg, Ina Hoeschele, David Herrington, Douglas A Bell, Yongmei Liu
Circulation. 2014;130:A16656
Abstract
Activation of the aryl-hydrocarbon receptor (AhR) promotes atherosclerosis in animal models and up-regulates the AhR repressor (AHRR). Intriguingly, the strongest and most robust epigenetic modifications reported in smokers are altered DNA methylation profiles of AHRR; however, the functional relevance of these modifications is unclear. Here we integrate AHRRmethylation profiles of monocytes (542 CpG sites ± 150kb of AHRR, using Illumina 450K array) with smoking status and ultrasound measured carotid plaque levels from 1,264 participants of the Multi-Ethnic Study of Atherosclerosis (MESA), as well as cis-gene expression profiles (± 1MB, using RNA sequencing) in a subset (373) of the monocyte samples. Association analysis was adjusted for age, sex, race, study site, and residual sample contamination.
RESULTS: Current smoking was associated with methylation of 34 AHRR sites (false discovery rate, FDR<0.01), with hypomethylation of cg05575921 as the top signal (FDR=2.7×10-131) as previously reported. Over half (59%) of the smoking-associated methylation profiles also correlated with cis-gene expression (of AHRR, EXOC3, or C5orf55, FDR<0.01). However, only AHRR expression was strongly associated with current smoking (p=3.3×10-21). Of the 18 AHRR-associated CpG sites, cg05575921 was most strongly correlated with AHRR expression (corr: -0.42, FDR=4.3×10-15). Cg05575921 methylation profiles also significantly mediated (p=9.1×10-6) the association between current smoking and higher carotid plaque levels. The associations of cg05575921 methylation with smoking (p=0.002) and atherosclerosis (extended fatty streaks, p=0.002) replicated in hepatic biopsies of 141 males (PDAY study). Further characterizing this region, we identified seven CpG sites within 180bp (using RRBS) with methylation correlated with cg05575921, which overlap many predicted regulatory features (in ENCODE and BLUEPRINT monocyte data).
CONCLUSIONS: Integration of DNA methylation and RNA sequencing data from ex vivomonocytes revealed a link between the smoking-related methylation of AHRR, induction of AHRR expression, and atherosclerosis. These findings also provide evidence for involvement of the AhR signaling pathway in smoking-related atherogenesis.
Article Information
vol. 130 no. Suppl 2 A16656
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Lindsay M Reynolds1;
- Ma Wan2;
- Jingzhong Ding3;
- Jackson R Taylor4;
- Kurt Lohman5;
- R Graham Barr6;
- Timothy D Howard1;
- Dan Su7;
- Devin Porter2;
- Ryan Gimple2;
- Gary S Pittman2;
- David Siscovick8;
- Bruce M Psaty9;
- Steven Shea10;
- David R Jacobs11;
- Stephen S Rich12;
- James E Hixson13;
- James H Stein14;
- Hendrik G Stunnenberg15;
- Ina Hoeschele16;
- David Herrington17;
- Douglas A Bell2;
- Yongmei Liu1
- 1Epidemiology and Prevention, Wake Forest Sch of Medicine, Winston-Salem, NC
- 2National Institute of Environmental Health Sciences, National Institute of Health, Rsch Triangle Park, NC
- 3Dept of Internal Medicine, Wake Forest Sch of Medicine, Winston-Salem, NC
- 4Dept of Gerontology and Geriatric Medicine, Wake Forest Sch of Medicine, Winston-Salem, NC
- 5Dept. of biostatistics, Div of Public Health Sciences, Wake Forest Sch of Medicine, Winston-Salem, NC
- 6Depts of Medicine and Epidemiology, Columbia Univ Med Cntr, New York, NY
- 7Epidemiology and Prevention, National Institute of Health, Rsch Triangle Park, NC
- 8New York Academy of Medicine, New York Academy of Medicine, New York, NY
- 9Cardiovascular Health Rsch Unit, Depts of Medicine and Epidemiology, Univ of Washington, Seattle, WA
- 10Dept of Epidemiology, Columbia Univ, New York, NY
- 11Div of Epidemiology and Community Health, Sch of Public Health, Univ of Minnesota, Univ of Minnesota, Minneapolis, MN
- 12Cntr for Public Health Genomics, Univ of Virginia, Charlottesville, VA
- 13Human Genetics Cntr, Sch of Public Health, Univ of Texas Health Science Cntr at Houston, Houston, TX
- 14Sch of Medicine and Public Health, Div of Cardiovascular Medicine, Univ of Wisconsin, Madison, WI
- 15Dept of Molecular Biology, Nijmegen Cntr for Molecular Life Sciences (NCMLS), Nijmegen, Netherlands
- 16Virginia Bioinformatics Institute, Virginia Tech, Blacksburg, VA
- 17Section on Cardiology, Wake Forest Sch of Medicine, Winston-Salem, NC
Abstract 13293: Acyl-coa Binding Protein is Marker of Myocardial Ischemia
Xu Shi, Gregory Lewis, Michelle Keyes, Laurie Farrell, Steven Carr, Hasmik Keshishian, Michelle O’Donoghue, Marc Sabatine, Robert Gerszten
Circulation. 2014;130:A13293
Abstract
Background: We recently applied a liquid chromatography tandem mass spectrometry (LC-MS/MS) based proteomics platform to plasma samples from individuals undergoing a “planned” myocardial infarction (PMI; alcohol ablation for hypertrophic cardiomyopathy) and identified acyl-CoA binding protein (ACBP) as a potential circulating biomarker of myocardial injury. ACBP is a 10 kDa intracellular protein that is highly expressed in cells with a high turn-over of fatty acids such as cardiomyocytes. We sought to determine the site of ACBP release and whether ischemia alone is sufficient to trigger an increment in plasma levels in humans.
Methods: Coronary sinus (CS) and peripheral venous samples were obtained simultaneously from PMI patients; peripheral venous samples were obtained from derivation/validation cohorts of subjects undergoing cardiac stress testing. Plasma ACBP levels were measured by immunoassay.
Results: In PMI patients (n=11), pre-injury levels of ACBP were comparable in the CS and peripheral samples. As early as 10 min after PMI, ACBP levels were 27% higher in the CS than in the periphery (P = 0.01), and remained 30% higher at 60 minutes (P = 0.02). Next, we studied 105 patients undergoing cardiac stress testing, 52 of whom demonstrated inducible ischemia (cases) and 53 of whom did not (controls). Baseline ACBP levels were comparable in cases and controls. Changes in median ACBP levels after the exercise stress challenge were significantly greater in the ischemic patients (cases) than in the controls (22% increase in cases; 1% decrease in controls; P = 0.0001), findings which were validated in a second cohort of 101 patients subjects (P = 0.002). These findings remained significant in a multivariable model adjusting for age, male sex, diabetes, BMI, smoking, creatinine, hypercholesterolemia, and total minutes of exercise. By contrast, the effects of exercise on established biomarkers (cTnI, NT-proBNP and FABP) did not differ betwen cases and controls.
Conclusions: We provide initial verification that ACBP is rapidly released from the injured heart and increases in response not only to infarction but also ischemia. These findings motivate additional clinical studies of this novel biomarker in heterogeneous patient cohorts.
Article Information
vol. 130 no. Suppl 2 A13293
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Xu Shi1;
- Gregory Lewis1;
- Michelle Keyes1;
- Laurie Farrell1;
- Steven Carr2;
- Hasmik Keshishian2;
- Michelle O’Donoghue3;
- Marc Sabatine3;
- Robert Gerszten1
- 1Cardiology, Massachusetts General Hosp, Boston, MA
- 2Cardiology, Broad Institute, Cambridge, MA
- 3Cardiology, Brigham and Women’s Hosp, Boston, MA
Abstract 11247: The Long Noncoding MALAT1 – MascRNA System is a Novel Regulator of Cardiac Innate Immunity
Wolfgang Poller, Martina Gast, Blanche Schroen, Antje Voigt, Jan Haas, Anna Papageorgiou, Uwe Kuehl, Dirk Lassner, Xiaomin Wang, Madlen Loebel, Sabrina Wilk, Nadine Althof, Tim Peters, Kannanganattu Prasanth, Hugo Katus, Benjamin Meder, Shinichi Nakagawa, Carmen Scheibenbogen, Stephane Heymans
Circulation. 2014;130:A11247
Abstract
Background: Regarding disease pathogenesis, it has become evident that confinement to analysis of protein-coding regions of the genome is insufficient since many noncoding regions are associated with human diseases. We searched for elements of the noncoding genome influencing the highly variable course of a cardiomyopathy caused by a common single-stranded RNA virus (CVB3).
Methods and Results: Transcriptome mapping of human CVB3 cardiomyopathy hearts showed high long noncoding RNAs (lncRNA) MALAT1 associated with spontaneous virus clearance and recovery, and low MALAT1 with virus persistence and clinical deterioration. Primary [[Unable to Display Character: ]]7kb MALAT1 transcript is only part of a complex RNA processing system generating e.g. mascRNA, a tRNA-like molecule, by a novel biosynthetic pathway. First, we found cell type-specific MALAT1 processing to result in grossly different mascRNA levels, which were highest in leukocytes. Second, CVB3 infection of genetically MALAT1-deficient (MALAT1-/-) mice resulted in anomalous splenic immune cell subtype distribution and function, and altered splenic and cardiac transcriptomes involving chemokine/receptor systems and antiviral genes. Third, recombinant mascRNA overexpression in primary cardiomyocytes induced IFIT genes coding an antiviral protein complex addressing 5’-triphosphate and 2’-O methylated RNA, and IFITM genes inhibiting multiple RNA viruses. Functionally, recombinant mascRNA blocked CVB3 replication in cardiomyocytes, inducing IFITs and IFITMs and immunoproteasome subunits, whereas the common time-delayed induction of interferons after infection was lacking.
Conclusions: These data indicate that the MALAT1-mascRNA system is a novel regulator of cardiac innate immunity influencing heart-immune system interactions e.g. during viral infections. mascRNA mimics or pharmacological modulation of the MALAT1-mascRNA system may have therapeutic potential.
Article Information
vol. 130 no. Suppl 2 A11247
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Wolfgang Poller1;
- Martina Gast1;
- Blanche Schroen2;
- Antje Voigt3;
- Jan Haas4;
- Anna Papageorgiou5;
- Uwe Kuehl1;
- Dirk Lassner1;
- Xiaomin Wang1;
- Madlen Loebel6;
- Sabrina Wilk6;
- Nadine Althof3;
- Tim Peters2;
- Kannanganattu Prasanth7;
- Hugo Katus4;
- Benjamin Meder4;
- Shinichi Nakagawa8;
- Carmen Scheibenbogen6;
- Stephane Heymans2
- 1Cardiology and Pneumology CBF, Charite – Universitätsmedizin Berlin, Berlin, Germany
- 2Cntr for Heart Failure Rsch, Cardiovascular Rsch Institute Maastricht (CARIM), Maastricht, Netherlands
- 3Biochemistry, Charite – Universitätsmedizin Berlin, Berlin, Germany
- 4Cardiology, Otto-Meyerhof-Centrum and Medizinische Klinik, Univ of Heidelberg, Heidelberg, Germany
- 5Cntr for Heart Failure Rsch, Cardiovascular Rsch Institute Maastricht (CARIM), Maastricht, Germany
- 6Immunology, Charite – Universitätsmedizin Berlin, Berlin, Germany
- 7Dept of Cell and Developmental Biology, Univ of Illinois at Urbana-Champaign, Urbana-Champaign, IL
- 8RNA Biology Laboratory, RIKEN Advanced Rsch Institute, Wako, Saitama, Japan
Abstract 19944: A PDE5A Gene Mutation Affecting Risk of Myocardial Infarction
Tan An Dang, Ingrid Braenne, Redouane Aherrahrou, Stephanie Tennstedt, Thorsten Kessler, Christian Hengstenberg, Jeanette Erdmann, Heribert Schunkert
Circulation. 2014;130:A19944
Abstract
INTRODUCTION: Multiple frequent genetic variants were shown to affect myocardial infarction (MI) risk. Genetic causes for familial clustering of MI are less clear. We aimed to identify and characterize the molecular underpinnings of premature MI in a family with 9 affected individuals.
METHODS AND RESULTS: Employing cosegregation analysis and exome sequencing we identified a mutation in the phosphodiesterase 5A (PDE5A) gene in all affected individuals (LOD score 3.16). It is located in an alternative promoter site of PDE5A and leads to a premature stop codon in one of the PDE5A isoforms (p.Lys7Ter). PDE5A encodes for three isoforms catalyzing cGMP, a second messenger mediating vasodilation and platelet passivation. Effects of the stop codon were investigated by western blot analysis after in vitromutagenesis. Overexpression in HEK cells did not reveal a loss of transcript but the expression of a N-terminally truncated protein. Deeper analyses of translation initiation by deletion of possible transcription starts via in vitro mutagenesis uncovered a protein lacking 91 amino acids compared to the full-length isoform. Activity of the truncated PDE5A was measured using PDEGlo (Promega). Moreover, the effect of the variant on the alternative promoter site was analyzed by luciferase assays. Therefore, a 600 bp fragment containing either the mutated or WT allele was cloned into a pGL4.10 vector (Promega). Overexpression in HEK cells showed 40% increase of promoter activity with the mutated allele (p<0.05). Similar results could be shown in other cell lines.
CONCLUSION: We identified a mutation in the PDE5A gene associated with premature MI. While a gain of function of PDE5A makes sense from a pathophysiological stance, the particular variant seemed to result in a premature stop codon. However, we could demonstrate that the variant might lead to increased promoter activity and that the presumable stop codon does not result into a loss of transcript but rather a truncated, potentially more active PDE5A isoform. Along with our homology modeling results and x-ray crystallographic and biochemical studies of Wang et al. (2010), these data support the idea of overexpression of a truncated though functional PDE5A in mutation carriers, possibly resulting in a gain function.
Article Information
vol. 130 no. Suppl 2 A19944
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Tan An Dang1;
- Ingrid Braenne2;
- Redouane Aherrahrou2;
- Stephanie Tennstedt2;
- Thorsten Kessler1;
- Christian Hengstenberg1;
- Jeanette Erdmann2;
- Heribert Schunkert1
- 1Klinik fuer Herz- und Kreislauferkrankungen, Deutsches Herzzentrum Muenchen, Munich, Germany
- 2Institute for Integrative and Experimental Genomics, Universitaet zu Luebeck, Luebeck, Germany
Abstract 19285: Notch1 Haploinsufficiency Increases Risk of Congenital Heart Defects in the Setting of Maternal Diabetes by an Epigenetic Mechanism
Madhumita Basu, Kevin Bosse, Vidu Garg
Circulation. 2014;130:A19285
Abstract
Congenital heart disease (CHD) is the most common type of birth defect. Epidemiologic studies have shown the importance of genetic and environmental factors in the multifactorial etiology of CHD. Maternal diabetes mellitus (DM) is one of the non-genetic risk factors that predisposes to CHD predominantly cardiac septation and cardiac outflow tract defects. DM is known to be associated with endothelial cell dysfunction and we recently demonstrated a genetic interaction between endothelial nitric oxide synthase and Notch1, which encodes a receptor that functions in an important cardiac developmental signaling pathway. We hypothesized that maternal DM in the setting of Notch1 heterozygosity of the developing embryo will predispose to CHD. Notch1+/- embryos (E13.5) exposed to maternal DM demonstrated an increased incidence (86%) of ventricular septal defects compared to wildtype littermates (22%) (Table). Gene expression studies in non-diabetic wildtype, diabetic wildtype and Notch1+/- embryos showed DM was associated with decreased Notch1 mRNA levels and upregulation in Jarid2, a histone H3K9 demethylase known to regulate Notch1. In H9C2 and endocardial-derived cells and chick embryos, we showed that hyperglycemia led to decreased expression of Notch1 and its downstream targets in a dose-dependent manner. Similarly, Jarid2 mRNA levels increased with high glucose. Furthermore, we found reduced luciferase reporter activity in cells transfected with a constitutively active Notch1 intracellular domain with hyperglycemia. Studies demonstrating the relative enrichment of Jarid2 on Notch1 locus with hyperglycemia by ChIP-qPCR will also be presented. Our findings reveal that maternal DM disrupts cardiac development by deregulating the Notch1 signaling pathway and suggest that this gene-environment interaction is mediated by an epigenetic mechanism involving Jarid2 providing the first mechanistic insights for this association.
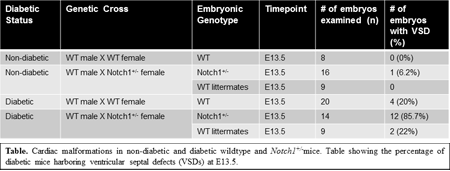
Article Information
vol. 130 no. Suppl 2 A19285
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Madhumita Basu;
- Kevin Bosse;
- Vidu Garg
- Cardiovascular and Pulmonary Rsch, Nationwide Children’s Hosp, Columbus, OH
Abstract 18946: A Complex Cardiac Rhythm and Conduction Abnormalities in Three Families Carrying a SCN5A Missense Mutation May Involve Single Nucleotide Polymorphisms in SCN5A and RYR2 genes
Erika Antunez-Arguelles, Argelia Medeiros-Domingo, Teresa Villarreal, Jonathan J Hernandez, Pedro Iturralde, Hector H Valdivia, Jonathan C Makielski, Carmen R Valdivia
Circulation. 2014;130:A18946
Abstract
Background: Mutations in the gene encoding the cardiac sodium channel (SCN5A) have been implicated in arrhythmia syndromes including Brugada (BrS) and long-QT type 3 syndromes. Also, wide spectrums of mixed phenotypes, known as “overlap syndromes”, have been associated to SCN5A mutations. The contributions of single nucleotide polymorphisms (SNPs) add more complexity to the disease phenotype. Here we report a missense mutation in SCN5A (T1708N) in three Hispanic families with history of sudden cardiac death (SCD) and extensive variability and severity of symptoms, including syncope, atrial and ventricular tachycardia, and conduction abnormalities.
Methods and Results: We used the Ion Torrent platform and AmpliSeq panel to sequence 19 genes associated with arrhythmia syndromes. In addition to SCN5A-T1708N mutation, the patients carry two common SNPs, SCN5A-H558R and the cardiac ryanodine receptor RYR2-Q2958R, which we previously reported to be highly prevalent (~30%) in Hispanics and to increase the risk of SCD in homozygous individuals. To study the functional properties of SCN5A-T1708N, the mutation was engineered by site-directed mutagenesis into SCN5A with or without the SCN5A-H558R and expressed in HEK cells for voltage clamp experiments. SCN5A-T1708N showed a significant increase of late INa compared to WT. Also, the kinetic properties of SCN5A-T1708N showed slower time constants of recovery and increase in slow inactivation, which would tend to reduce INa; these effects were not modified by the presence of SCN5A-H558R. On the other hand, INa density for SCN5A-T1708N was reduced compared to WT (289± 63 and 387± 62 pA/pF, respectively), yet, for the double mutant H558R-T1708N, INa was more severely depressed (145± 32 pA/pF).
Conclusion: The profound biophysical phenotype of T1708N with loss and gain of function could account for the variability and the severity of the clinical phenotype in these families. The RYR2-Q2958R polymorphism may function as a pro-arrhythmic genetic modifier and it is conceivable that it may predispose to SCD in some members of these families. The study of these polymorphisms in large cohorts may identify common haplotypes associated to cardiac arrhythmias and facilitate identification of patients at high risk for SCD.
Article Information
vol. 130 no. Suppl 2 A18946
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Erika Antunez-Arguelles1;
- Argelia Medeiros-Domingo2;
- Teresa Villarreal3;
- Jonathan J Hernandez1;
- Pedro Iturralde4;
- Hector H Valdivia1;
- Jonathan C Makielski5;
- Carmen R Valdivia1
- 1Internal Medicine, Univ of Michigan, Ann Arbor, MI
- 2Arrhythmias, Cardiogenetics, INSEArrhythmias, Cardiogenetics LSPITAL, Bern, Switzerland
- 3Laboratorio de Genómica de las Enfermedades Cardiovasculares, INMEGEN, Mexico City, Mexico
- 4Internal Medicine, Univ of Michigan, Mexico City, Mexico
- 5Medicine, Univ of Wisconsin, Madison, WI
Abstract 18455: Genome Editing Identifies a Functional Role for the Lead Variant at the ZC3HC1 Coronary Artery Disease Risk Locus in Cell Cycle Regulation
Peter D Jones, Mike Kaiser, Thomas R Webb, Nilesh J Samani
Circulation. 2014;130:A18455
Abstract
Genome wide association studies have identified 46 chromosomal loci that are associated with coronary artery disease (CAD); however, for most of these loci the mechanism by which they affect CAD risk is unclear. The association signal at chromosome 7q32.2 is rare amongst CAD GWAS loci as the lead variant, rs11556924, introduces a coding change (His363Arg) in the ZC3HC1 gene. No other variants are in strong linkage disequilibrium, suggesting that this is the functional SNP. During interphase, ZC3HC1 degrades Cyclin B1, keeping its levels low. Inactivation of ZC3HC1 and subsequent accumulation of Cyclin B1 triggers entry into mitosis. We hypothesised that the effect of the SNP perturbs the function of ZC3HC1 in regulating the cell cycle. To investigate this, we used recombinant adeno-associated virus mediated genome editing to generate isogenic DLD-1 cell lines that are identical apart from at rs11556924. A proliferation assay comparing the rate of growth of cells of the homozygous non-risk and heterozygous isogenic lines showed a decrease in cellular growth in cells carrying one copy of the risk allele, with a mean of 25.4% fewer cells after 5 days growth (p=0.019). Counter-intuitively, cells carrying the risk allele also showed an increase in mitotic index, with the number of cells in mitosis after 2 hours of mitotic arrest induced with the microtubule-depolymerising drug colchicine, increasing from μ=7.25% (S.E.=0.71%) to μ=12.32% (S.E.=1.71%) (p=0.0001). This increase in mitotic index could be caused by chromosome segregation errors that may be causing cells to arrest in mitosis, which would be expected to result in a widening in the distribution of chromosome numbers in these cells. We tested this by counting the number of chromosomes in cells carrying the risk allele compared to those which are homozygous for the non-risk allele but found no difference in chromosome number (p=0.78). These data suggest that the CAD risk allele of rs11556924 perturbs Cyclin B1 dynamics causing a delay in the progression of mitosis. The mechanism by which this increases CAD risk requires further elucidation.
Article Information
vol. 130 no. Suppl 2 A18455
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Peter D Jones;
- Mike Kaiser;
- Thomas R Webb;
- Nilesh J Samani
- Dept of Cardiovascular Sciences, Univ of Leicester, Leicester, United Kingdom
Abstract 18575: The Transcriptional Cofactor Eyes Absent 4 is a Critical Regulator for Maintaining Normal Cardiac Function
Tatjana Williams, Moritz Hundertmark, Peter Nordbeck, Sabine Voll, Anahi Paula Arias-Loza, Ines Elsner, Daniel Oppelt, Silvana Olivares, Jost Schoenberger, Oliver Ritter
Circulation. 2014;130:A18575
Abstract
Introduction: E193, a human mutation in the transcription cofactor Eyes absent 4 (EYA4) causes hearing impairment followed by terminal heart failure, defining an important role for Eya4 in maintaining normal cardiac function.
METHODS AND RESULTS: First, in-vitro experiments show that overexpression of Eya4 and the mutant isoform alter the expression of p27kip1 on both, transcript and protein levels. Next, we generated transgenic mice with cardiomyocyte-specific Eya4 or E193 overexpression to elucidate the in vivo function of Eya4 upon cardiac physiology. Luciferase and CHIP assays revealed that Eya4 and E193 bind to and regulate p27 expression in a contradictory manner, as already seen in vitro. Activity and phoshorylation of the downstream molecules CK2α and HDAC2 were significantly elevated in Eya4 mice, whereas they were significantly reduced in E193 animals compared to WT littermates. MRI and hemodynamic analysis indicate that a constitutive overexpression of Eya4 results in the age-dependent development of hypertrophy already under baseline conditions with no obvious functional effects, whereas E193 overexpressing animals develop onset of dilative cardiomyopathy as seen in human patients carrying the E193 mutation. Morphometric analysis proved ventricular hypertrophy or dilation of the LV associated with a thinning of the myocardial wall, interstitial fibrosis of myocardial tissue and alterations in cell size. Re-activation of fetal genes also occured in both TG models, characteristic for cardiac disease. Both cardiac phenotypes were aggravated upon pressure overload.
Finally, we identified a new human heterozygous truncating Eya4 mutation, E215, which leads to similar clinical features of disease and a stable myocardial expression of the mutant protein.
Conclusion: Our results implicate that Eya4 plays a critical role in regulating normal cardiac physiology and function via Six1/p27/CK2α/HDAC2 and that an imbalance within the Eya4/Six1 transcriptional complex leads to an age dependent onset of cardiomyopthy and heart failure.
Article Information
vol. 130 no. Suppl 2 A18575
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Abstract 15694: Absence of ECM Remodeling in Smad3 Mutant Mice Leads to Aggressive and Accelerated Aneurysmal Growth Through Distorted Downstream TGF-β Signaling
Ingrid van der Pluijm, Nicole van Vliet, Luna Buijs-Offerman, Aida Bertoli-Avella, Jeroen Essers
Circulation. 2014;130:A15694
Abstract
Aneurysm-osteoarthritis syndrome (AOS), caused by SMAD3 gene mutations, is an autosomal dominant condition characterized by aortic aneurysms and early-onset osteoarthritis. As part of the transcription factor complex Smad2/3/4, Smad3 is essential for TGFβ-activated downstream transcription of CTGF, MMPs, SMAD7 and others. Smad3-/-mice show aneurysmal development at young age, but the underlying mechanism is unknown. To better understand the processes involved, we performed thorough phenotypic and molecular analyses of aneurysmal growth in Smad3-/- mice. First, echocardiograms of cross-sectional studies in Smad3-/- mice showed a significant increase in diameter of root and ascending aorta (18-20%), and significant increased aortic length (16-20%) already at 6 weeks of age, but no difference in aortic distensibility. Importantly, 50% of Smad3-/- mice died suddenly between age 6-24 weeks. Successive macroscopic analysis showed up to a 5-fold increase in aortic diameter of the ascending aorta. Next, longitudinal studies showed a steep increase of aneurysmal growth within only 6 weeks. As the aneurysmatic aortic wall remained translucent, this was indicative for the absence of large scale extracellular matrix remodeling or collagen deposition. Indeed, immunohistochemistry showed a disturbed vascular wall and major sites without elastin or collagen. In accordance, in vivo molecular imaging showed increased aortic neutrophil elastase activity. Remarkably, CT-scans showed dilatations of the arterial tree at different sites of the body, as in AOS patients. In absence of Smad3, we still observed increased nuclear translocation of its co-transcription factor (p)Smad2, and upregulated pERK signaling, inferring increased upstream TGFβ signaling. Hence we reasoned that Smad3 deficiency results in activated TGFβ signaling due to inability to activate downstream Smad7 transcription, a potent inhibitor of TGFβ signaling. Indeed, downstream TGFβ signaling seemed abolished as derived from the absence of MMP activation in Smad3-/- mouse aortas. Identifying the underlying molecular mechanism due to Smad3 mutations is crucial to provide the correct ‘personalized’ treatment for aneurysmal disease.
Article Information
vol. 130 no. Suppl 2 A15694
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Ingrid van der Pluijm1;
- Nicole van Vliet2;
- Luna Buijs-Offerman3;
- Aida Bertoli-Avella3;
- Jeroen Essers4
- 1Vascular Surgery, Erasmus Med Cntr, Rotterdam, Netherlands
- 2Genetics, Erasmus Med Cntr, Rotterdam, Netherlands
- 3Clinical Genetics, Erasmus Med Cntr, Rotterdam, Netherlands
- 4Vascular Surgery, Genetics, Radiation Biology, Erasmus Med Cntr, Rotterdam, Netherlands
Abstract 15042: Arrhythmogenic Cardiomyopathy Mutations Cause Disassembly of the Cx43 Forward Trafficking Machinery Which Can Be Rescued by GSK-3β Inhibition
Shan-Shan Zhang, Dagmar A Kuhn, Elise Kessler, Angeliki Asimaki, Valentin Sottas, Dimitri Vanhecke, Barbara Rothen-Rutishauser, Jeffrey E Saffitz, Robin M Shaw, Andre G Kleber
Circulation. 2014;130:A15042
Abstract
Arrhythmogenic cardiomyopathy leads to a decrease in connexin43 (Cx43) immunosignal at the intercalated disc, despite unchanged total Cx43 protein. In an experimental model of arrhythmogenic cardiomyopathy, involving transfection of neonatal ventricular cardiomyocytes and HeLa cells with mutated plakoglobin (2057del2 plakoglobin, corresponding to Naxos disease in humans), we quantified the decrease in Cx43 at the intercalated disc by morphometric analysis of confocal slices, and analyzed the EB1-dependent microtubular trafficking machinery known to be responsible for forward trafficking of Cx43 cargo to the intercalated disc. Expression of mutated plakoglobin reduced Cx43 signal at the intercalated disc from 100±13% to 47±2%, n=9 (mean±S.E). In HeLa cells, a cell line known to express the full EB1-dependent trafficking machinery, mutated plakoglobin produced a marked disassembly of microtubules, characterized by a failure of alignment of microtubules towards the cell border, less developed EB1 protein comets at microtubule plus-ends, and a lack of a discernable microtubule organizing center (MTOC). Glycogen synthase kinase-3β (GSK-3β) is a known constitutive inhibitor of microtubule dynamics and EB1 activity. SB216763, a specific inhibitor of GSK-3β fully rescued the MTOC, microtubular alignment and EB1 activity at microtubule plus-ends.
These results suggest that decreased connexin43 at the cardiac cell-to-cell border in arrhythmogenic cardiomyopathy patients is attributable to disassembly of the microtubule based forward trafficking machinery. Furthermore, inhibition of GSK-3β, rescues both EB1 activity and microtubule dynamics to properly localize Cx43 at cell-to-cell borders.
Article Information
vol. 130 no. Suppl 2 A15042
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Shan-Shan Zhang1;
- Dagmar A Kuhn2;
- Elise Kessler3;
- Angeliki Asimaki4;
- Valentin Sottas4;
- Dimitri Vanhecke2;
- Barbara Rothen-Rutishauser2;
- Jeffrey E Saffitz4;
- Robin M Shaw5;
- Andre G Kleber4
- 1Heart Institute and Dept of Medicine, Cedars-Sinai Med Cntr, Heart Institute, Cedars-Sinai Med Cntr, Los Angeles, CA
- 2Adolphe Merkle Institute, Fribourg Univ, Marly, Switzerland
- 3Dept of Pathology, BIDMC, Harvard Med Sch, Boston, MA
- 4Pathology, BIDMC, Harvard Med Sch, Boston, MA
- 5Heart Institute and Dept of Medicine, Heart Institute, Cedars-Sinai Med Cntr, Los Angeles, CA
Abstract 13801: The p.Leu167del Mutation in APOE Gene causes Autosomal Dominant Hypercholesterolemia by Down-Regulation of LDL Receptor Expression in Hepatocytes
Ana Cenarro, César Martín, Marianne Stef, Isabel de Castro-Orós, Aitor Etxebarria, Lourdes Palacios, Rocío Mateo-Gallego, Helena Ostolaza, Teresa Tejedor, Fernando Civeira
Circulation. 2014;130:A13801
Abstract
Introduction: The p.Leu167del mutation in APOE gene has been associated to different hyperlipidemias. However, the frequency of this mutation in autosomal dominant hypercholesterolemia and the mechanism of this association is not known.
Objectives: To establish the frequency of p.Leu167del mutation in APOE gene in subjects with clinical diagnosis of Autosomal Dominant Hypercholesterolemia (ADH) non-dependent of LDLR, APOB nor PCSK9 genes, and to investigate the mechanism by which this mutation associates to ADH.
Methods: We selected 288 unrelated subjects with ADH non-dependent of LDLR, APOB nor PCSK9 genes, and 220 unrelated normolipidemic subjects (control group). All available family members of mutation carrier subjects were also studied. Exon 4 of APOE gene was sequenced in all subjects.
VLDL and LDL were isolated from p.Leu167del carriers and E3/E3 control subjects by ultracentrifugation. Quantification of lipoprotein uptake in HepG2 and THP-1 cells and LDL receptor membrane-expression in HepG2 cells were carried out by flow cytometry.
Results: In the ADH group, 9 unrelated subjects (3.1%) were carriers of the p.Leu167del mutation in APOE gene and none in the control group. Eleven family members were carriers of the mutation, 7 with isolated hypercholesterolemia and 4 with mixed hyperlipidemia. A clear co-segregation of p.Leu167del mutation with elevated plasma cholesterol levels in the families was found.
VLDL isolated from p.Leu167del mutation carriers had a significantly higher uptake by HepG2 and THP-1 cells compared to VLDL isolated from E3/E3 subjects (p<0.01). When increasing incubation times of HepG2 cells with VLDL from p.Leu167del carriers, the LDL receptor expression in surface membrane diminished, in contrast with the VLDL from E3/E3 subjects. When pre-incubating HepG2 cells with VLDL from p.Leu167del mutation carriers, the percentage of LDL internalisation was significantly lower than when incubating with VLDL from E3/E3 subject (p<0.01).
Conclusion: The p.Leu167del mutation in APOE gene is associated to ADH in 3.1% of studied subjects. The mechanism of this association is higher uptake of VLDL, down regulation of the LDLR expression and decrease of LDL internalisation in hepatocytes.
Article Information
vol. 130 no. Suppl 2 A13801
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Ana Cenarro1;
- César Martín2;
- Marianne Stef3;
- Isabel de Castro-Orós1;
- Aitor Etxebarria2;
- Lourdes Palacios3;
- Rocío Mateo-Gallego1;
- Helena Ostolaza4;
- Teresa Tejedor5;
- Fernando Civeira1
- 1Laboratorio de Investigación Molecular, Hosp Univ Miguel Servet, Zaragoza, Spain
- 2Bioquímica y Biología Molecular, UPV/EHU, Bilbao, Universidad del País Vasco, Bilbao, Spain
- 3Genomics, Progenika Biopharma, S.L., Vizcaya, Spain
- 4Bioquímica y Biología Molecular, UPV/EHU, Universidad del País Vasco, Bilbao, Spain
- 5Anatomía, Embriología y Genética, Universidad de Zaragoza, Zaragoza, Spain
Abstract 11923: FLNC Gene Splice Mutation Causes Dilated Cardiomyopathy in Two Families
Rene L Begay, August Martin, Sharon L Graw, Dobromir B Slavov, Charles A Tharp, Mary Sweet, Francesca Brun, Kenneth L Jones, Katherine Gowan, Daniela Miani, Gianfranco Sinagra, Luisa Mestroni, Deborah M Garrity, Matthew R Taylor
Circulation. 2014;130:A11923
Abstract
Background: Dilated cardiomyopathy (DCM) is an important and frequently genetic cause of heart failure. Over 30 DCM genes have been described, the majority of which encode proteins that contribute to cytoskeletal and sarcomeric structures. Only 30-40% of cases can be attributed to a known DCM gene, motivating the ongoing search for novel disease genes.
Methods and Results: We performed whole exome sequencing (WES) in a multigenerational DCM family from Northern Italy in whom prior DCM genetic testing had been unrevealing. Pathogenic variants were sought by a combination of bioinformatic filtering and cosegregation among affected individuals within the family. Thirteen gene candidates were identified including one novel variant in FLNC (filamin-C gene), already linked to a skeletal muscle disease phenotype. We further noted that WES found the identical variant in a second smaller family from the same geographical area; the mutation was present on a common haplotype. The variant is located in the 3’ end of the gene and is predicted to disrupt splicing and produce haploinsufficiency for the FLNC protein. Our patients showed no evidence of skeletal myopathy, previously implicated in FLNC mutations. In-situ hybridization demonstrated cardiac filamin expression in zebrafish and morpholino knockdown of zebrafish FLNC-b led to a heart failure phenotype in the zebrafish model.
Conclusion: Using WES, we have identified a novel gene, associated with DCM called FLNC. Furthermore, we have generated a zebrafish model that recapitulates the human FLNC DCM phenotype.
Article Information
vol. 130 no. Suppl 2 A11923
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Rene L Begay1;
- August Martin2;
- Sharon L Graw1;
- Dobromir B Slavov1;
- Charles A Tharp1;
- Mary Sweet1;
- Francesca Brun3;
- Kenneth L Jones4;
- Katherine Gowan4;
- Daniela Miani3;
- Gianfranco Sinagra3;
- Luisa Mestroni1;
- Deborah M Garrity2;
- Matthew R Taylor1
- 1Cardiovascular Institute and Adult Med Genetics Program, Univ of Colorado Denver – Anschutz Med Campus, Aurora, CO
- 2Biology, Colorado State Univ, Ft. Collins, CO
- 3Cardiovascular Dept, Univ of Trieste Hosp, Trieste, Italy
- 4Biochemistry and Molecular Genetics, Univ of Colorado Denver – Anschutz Med Campus, Aurora, CO
Abstract 11990: LMNA Cardiomyopathy Mimicking Arrhythmogenic Right Ventricular Cardiomyopathy
Koichi Kato, Seiko Ohno, Takeru Makiyama, Minoru Horie
Circulation. 2014;130:A11990
Abstract
Background and Objectives: Arrhythmogenic right ventricular cardiomyopathy (ARVC) is an inherited disease characterized by RV dilatation and ventricular arrhythmias. Desmosomal gene mutations are the major cause of ARVC. LMNA mutations have been known to lead dilated cardiomyopathy, other systemic diseases, and more recently, ARVC. In this study, we performed extensive genetic screening for LMNA in ARVC patients and assessed the clinical characteristics of patients with LMNA mutations.
Methods: Study cohort consisted of 57 ARVC probands (definite; 45). Coding exons of LMNA, 4 desmosomal protein genes (PKP2, DSP, DSG2, DSC), and also 3 long QT syndrome related genes (KCNQ1, KCNH2, SCN5A) were amplified and sequenced by using illumina next generation sequencer. Clinical characteristics of LMNA mutation carriers and those of desmosomal mutation carriers were compared by using student t test.
Results: Among 57 clinically-diagnosed ARVC probands, we identified desmosomal mutations in 32 probands (56.1%) and two LMNA mutations in two probands. The first LMNA mutation, p.M1K was detected in 62-year-old male, and the second one, p.W514X was in 70-year-old male. Both patients showed RV dilatation, non-sustained ventricular tachycardia, and complete atrioventricular block. His younger brother also died from ARVC. The proband’s daughter and son, who are currently in their 30s, have the same M1K mutation, however, have not had any signs of ARVC yet. In the family member with W514X mutant, the proband’s father suddenly died in his 40s and 45-year-old daughter who had the same W514X mutation, showed RV dilatation and brady-AF. In probands with LMNA mutations compared to those with desmosomal mutations, the age of onset was significantly older (38.6±18.1 vs 60.0±2.8), and their heart rate was significantly slower (61.1±12.5 vs 47±1.4). Both probands with LMNA mutations underwent pacemaker therapy, which is rare in patients with desmosomal mutations (2/2 vs 1/32 ). In family members with LMNA mutations, none of mutation carriers had showed ARVC until their 50s.
Conclusion: Our patients with LMNA mutations developed ARVC with bradyarrhythmia after age of 50. Genetic screening for LMNA gene is important for ARVC, especially in cases with bradycardia.
Article Information
vol. 130 no. Suppl 2 A11990
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Dept of Cardiovascular and Respiratory Medicine, Shiga university of medical science, Otsu, Japan
- 2Dept of Cardiovascular Medicine, Kyoto Univ, Kyoto, Japan
Abstract 19297: Susceptibility Loci for Clinical CAD Predispose to Subclinical Coronary Atherosclerosis Throughout the Life Course
Elias Salfati, Shuktika Nandkeolyar, Stephen Fortmann, Stephen Sidney, Mark A Hlakty, Thomas Quertermous, Alan S Go, Carlos Iribarren, Benjamin A Goldstein, Themistocles L Assimes
Circulation. 2014;130:A19297
Abstract
Recent genome wide association studies (GWAS) have identified 49 single nucleotide polymorphisms (SNPs) associated with clinically significant complications of CAD including myocardial infarction (MI), CABG, PCI, and/or angina. The mechanism by which these loci influence the risk of clinical CAD remains largely unclear. We hypothesized that variants at these loci collectively facilitate the formation of coronary plaque in a monotonic fashion throughout the life course. We used genetic data from dbGAP (SEA, FHS, and MESA) as well as from the Stanford-Kaiser ADVANCE study imputed to the 1000 genomes project to examine the association between a genetic risk score (GRS) of high-risk alleles at these 49 SNPs and the presence of subclinical atherosclerosis. Subclinical atherosclerosis was identified by either pathologic examination of the coronary arteries or by radiographic assessment of coronary artery calcification (CAC). We stratified white/European subjects within each study into one of five age groups (≤30, 31-45, 46-60, 61-75, >75 years) and defined cases as subjects with either any raised lesions in their right coronary artery on autopsy (SEA, 26.7% subjects aged 18 to 30 years at time of unexpected death) or with an age and sex specific CAC score >75th percentile (all other studies, age > 30 years). Among 1561 cases and 5068 controls, we found a one SD increase in the GRS was associated with a 28% increased risk of having advanced subclinical coronary atherosclerosis (p = 3.82 x 10-16). This increase in risk was significant in every age stratum (.01 > p > 9.4 x 10-7)and was remarkably similar across all age strata (p test of heterogeneity = 0.99). We obtained near identical results and levels of significance when we restricted the GRS to 33 SNPs not associated with traditional risk factors. Our findings strongly support the notion that susceptibility alleles for clinical CAD uncovered through large-scale meta-analysis of GWAS uniformly promote the development of coronary atherosclerosis from birth. This predisposition is sustained at a constant level throughout one’s lifetime. Given it is observed at the earliest stage of plaque formation, it is unlikely to involve a concurrent predisposition to plaque rupture and/or thrombosis.
Article Information
vol. 130 no. Suppl 2 A19297
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Elias Salfati1;
- Shuktika Nandkeolyar2;
- Stephen Fortmann3;
- Stephen Sidney4;
- Mark A Hlakty1;
- Thomas Quertermous1;
- Alan S Go4;
- Carlos Iribarren4;
- Benjamin A Goldstein1;
- Themistocles L Assimes1
- 1Medicine, Stanford Univ Sch of Medicine, Stanford, CA
- 2Medicine, Med College of Wisconsin, Milwaukee, WI
- 3The Cntr of Health Rsch, Kaiser Permanente, Portland, OR
- 4Div of Rsch, Kaiser Permanente, Oakland, CA
Abstract 18767: Association of Protein-Coding Genetic Variants with Coronary Arterial Calcification in 21,000 Individuals of European and African Ancestries
Pradeep Natarajan, Lawrence F Bielak, Joshua C Bis, Donald Bowden, Mary F Feitosa, Vilmundur Guðnason, Shih-Jen Hwang, Sekar Kathiresan, Maryam Kavousi, Leo-Pekka Lyytikäinen, Hayato Tada, Jessica Van Setten, Lisa R Yanek, Jie Yao, Laura M Yerges-Armstrong, Christopher J O’Donnell
Circulation. 2014;130:A18767
Abstract
Introduction: Coronary arterial calcification (CAC) is a marker of subclinical atherosclerosis in asymptomatic individuals, and is a heritable risk factor that correlates with the risk of coronary artery disease (CAD) development. Genotyping protein-coding regions of the genome with an array is a scalable approach to potentially discover causal low-frequency and rare protein-coding mutations with large phenotypic effects.
Methods: Using the Illumina HumanExome BeadChip, we genotyped 247,870 variants (of which 231,539 are in exons) in each of 17,953 participants of European ancestry and 3,078 participants of African ancestry without clinical CAD from the Diabetes Heart Study, Framingham Heart Study, Family Heart Study, Cardiovascular Health Study, Rotterdam Study, Age Gene/Environment Susceptibility Study, Multi-Ethnic Study of Atherosclerosis, Dutch & Belgian Lung Cancer Screening Trial, Young Finns Study, Gene Study of Atherosclerosis Risk in Families, High Risk Plaque Bioimage Study, Old Order Amish Study, and Jackson Heart Study. We tested whether protein-coding variants, individually or aggregated within a gene, were associated with CAC, both as a continuous and binary variable.
Results: We first robustly replicated prior CAC genomic non-coding association signals at 9p21 (p=9.85 x 10-29) and PHACTR1 (p=4.10 x 10-23) identified in GWAS based on HapMap imputation. A rare nonsynonymous variant in APOB (rs5742904) was associated with increased CAC (p=1.01 x 10-10) although this signal was driven by the Old Order Amish where it is a known founder mutation. The nonsynonymous APOE rs7412 variant (maf=0.078), the APOE epsilon2 allele, was associated with reduced CAC (p=2.61 x 10-9). Additionally, there was evidence of association for the nonsynonymous SSTR3 rs4988466 variant (maf=0.046) (p=7.77 x 10-8). SSTR3 is involved in cell proliferation, among other functions. Gene-based burden approaches did not yield statistically significant associations.
Conclusions: We discovered nonsynonymous coding variants in APOB, APOE, and SSTR3 associated with CAC in a meta-analysis approach of 21,000 individuals using an exon-enriched genotyping array.
Article Information
vol. 130 no. Suppl 2 A18767
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Pradeep Natarajan1;
- Lawrence F Bielak2;
- Joshua C Bis3;
- Donald Bowden4;
- Mary F Feitosa5;
- Vilmundur Guðnason6;
- Shih-Jen Hwang7;
- Sekar Kathiresan1;
- Maryam Kavousi8;
- Leo-Pekka Lyytikäinen9;
- Hayato Tada10;
- Jessica Van Setten11;
- Lisa R Yanek12;
- Jie Yao13;
- Laura M Yerges-Armstrong14;
- Christopher J O’Donnell15,
- Cohorts for Heart and Aging Rsch in Genomic Epidemiology (CHARGE) Consortium
- 1Medicine, Cardiology, Massachusetts General Hosp, Boston, MA
- 2Sch of Public Health, Epidemiology, Univ of Michigan, Ann Arbor, MI
- 3Cardiovascular Health Rsch Unit, Univ of Washington, Seattle, WA
- 4Sch of Medicine, Biochemistry, Genomics, Personalized Medicine, Wake Forest, Winston-Salem, NC
- 5Sch of Medicine, Genetics, Washington Univ, St. Louis, MO
- 6Heart Preventive Clininc and Rsch Institute, Icelandic Heart Association, Kópavogur, Iceland
- 7Framingham Heart Study, Cntr for Population Studies, National Heart, Lung, and Blood Institute, Framingham, MA
- 8Epidemiology, Erasmus Univ Med Cntr, Rotterdam, Netherlands
- 9Sch of Medicine, Clinical Chemistry, Univ of Tampere, Tampere, Finland
- 10Emergency Medicine, Kanazawa Univ, Kanazawa, Japan
- 11Med Genetics, Univ Med Cntr Utrecht, Utrecht, Netherlands
- 12Sch of Medicine, General Internal Medicine, Johns Hopkins Univ, Baltimore, MD
- 13Med Genetics, Cedars Sinai Med Cntr, Los Angeles, CA
- 14Sch of Medicine, Program in Personalized and Genomic Medicine, Univ of Maryland, Baltimore, MD
- 15Framingham Heart Study, 2Cardiovascular Epidemiology and Human Genomics Branch, National Heart, Lung and Blood Institute, Framingham, MA
Abstract 16274: Identification of Novel CAD Genetic Loci by 1000 Genomes-Based Imputation and a Non-Additive Discovery Screen
Majid Nikpay, Anuj Goel, Hong-Hee Won
Circulation. 2014;130:A16274
Abstract
Introduction: Known common coronary artery disease (CAD) risk variants explain only 10% of the predicted genetic heritability of the disease, suggesting that important genetic signals remain to be discovered.
Hypothesis: The 1000 Genomes imputation training set, and non-additive discovery screens, may allow detection of additional CAD-associated genetic variants that contribute to missing heritability.
Methods: As part of the CARDIoGRAMplusC4D Consortium, we assembled 48 GWAS of CAD that included 1000 Genomes imputed data. These consisted of 60,801 CAD cases and 123,504 controls, 23% being of non-European ancestry. In each study, GWAS analysis was carried out by assuming an additive, dominant or recessive model of inheritance, and followed by meta-analysis to combine the GWAS results for each model.
Results: After QC filtering, 9.4 million variants (91% SNPs, 9% INDELs) were available for meta-analysis. 29% of these were lower frequency variants (0.005 < MAF < 0.05). Novel associations (P < 5 x 10-8, and outside of known CAD genomic regions) under the additive model were detected for 38 variants (8% INDELs) with imputation info score of 0.94 [0.88-0.96] (median [IQR]). Of note, 34% of novel variants (N=13) were of low allele frequency (MAF < 0.05; median [IQR] = 0.03 [0.02-0.03]); these exhibited much larger effect sizes (P < 0.0001; Cohen’s d = 2.3) as compared to the common variants (MAF ≥ 0.05). The newly identified variants were mapped to 10 novel genomic loci for CAD. Together, these variants explained 2.5% of the heritability of CAD and majority (60%) were intronic. Three of these loci fit a dominant mode of inheritance. An additional two novel loci were identified by a recessive mode of inheritance. Among the newly identified loci, three had been previously reported at GWAS levels of significance for metabolic traits.
Conclusions: These findings demonstrate the value of using a global imputation training set to enhance coverage of low allele frequency and incompletely tagged variants. Consideration of non-additive models of inheritance enabled identification of additional genetic variants associated with CAD.
Article Information
vol. 130 no. Suppl 2 A16274
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Ruddy Canadian Cardiovascular Genetics Cntr, Univ of Ottawa Heart Institute, Ottawa, Canada
- 2Cardiovascular Medicine. Radcliffe Dept of Medicine, Univ of Oxford, Oxford, United Kingdom
- 3Cntr for Human Genetic Rsch, Massachusetts General Hosp, Boston, MA
Abstract 12291: Contribution of Rare Mutations in Mendelian Hypercholesterolemia Genes to Risk for Premature Coronary Artery Disease in the Population
Hong-Hee Won, Ron Do
Circulation. 2014;130:A12291
Abstract
Introduction: Low-density lipoprotein cholesterol (LDL-C) is a causal risk factor for coronary artery disease (CAD). Rare mutations in at least 6 genes lead to Mendelian forms of high or reduced LDL-C; three (APOB, LDLR, PCSK9) act in a dominant pattern whereas three (ABCG5, ABCG8, LDLRAP1) in a recessive pattern. We address to what extent rare mutations in Mendelian LDL-C genes contribute to early CAD risk in the population.
Methods: We sequenced the exons of the 6 genes in 9,329 early CAD cases (myocardial infarction, angiographic CAD, or coronary revascularization in men≤50 and women≤60) and 10,245 controls from 9 studies using targeted and whole exome next-generation sequencing. We tested 3 sets: ‘Null alleles’ (nonsense, splice-site, or frameshift); ‘Deleterious (7/7)’ (null and missense annotated as damaging by 7 algorithms); and ‘Deleterious (6/7)’ (null and missense annotated as damaging by at least 6 algorithms). Given the rarity of deleterious mutations, we aggregated these mutations in each gene and tested for an excess or deficit in cases vs. controls.
Results: Counts of mutations are provided in Table. Null mutations in LDLR, carried by 1:500 participants, confered a 8-fold increase in CAD risk (P=8х10-7) whereas heterozygosity for a null mutation in ABCG5 (1:650 frequency) was associated with a 3-fold increased risk (P=5х10-3). ‘Deleterious (7/7)’ mutations in LDLR, carried by 1:100 participants, confered a 4-fold increased risk (P=8х10-17) whereas heterozygosity for a ‘Deleterious (7/7)’ mutation in ABCG5 (1:250 frequency) was associated with a 2-fold increased risk (P=2х10-3). Heterozygous null allele carriers at LDLR and ABCG5 had increased LDL-C (P<0.001).
Conclusions: Of early CAD cases, 2-3% carry a rare, deleterious mutation at LDLR or ABCG5 associated with increased risk. Although previously reported to cause recessive sitosterolemia, we find that heterozygosity for a null allele at ABCG5 is associated with markedly higher early CAD risk.
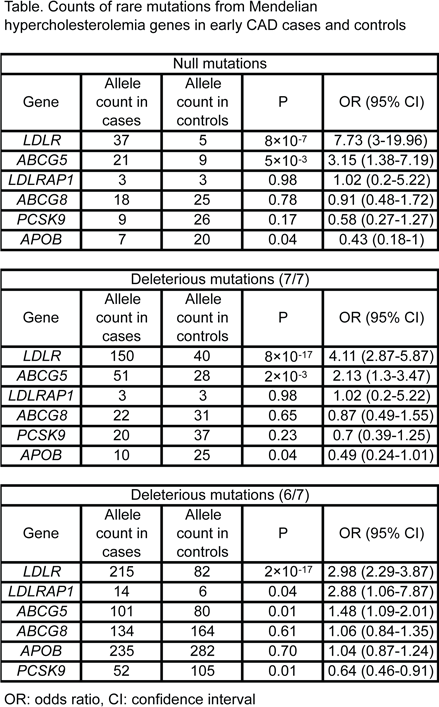
Article Information
vol. 130 no. Suppl 2 A12291
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Hong-Hee Won;
- Ron Do,
- on behalf of the Myocardial Infarction Genetics Sequencing Consortium
- Cntr for Human Genetic Rsch, Massachusetts General Hosp, Boston, MA
Abstract 19836: Lipid Metabolism is Modified by the Interaction Between Cholesteryl Ester Transfer Protein Gene Polymorphism and Mediterranean Diet in Patients in Secondary Prevention for Cardiovascular Disease
Antonio Garcia Rios, Francisco Gomez-Delgado, Ana Isabel Perez Caballero, Andreea Corina-Baba, Vanesa Navarro-Martos, Carmen Marin-Hinojosa, Jose Lopez-Miranda, Pablo Perez-Martinez
Circulation. 2014;130:A19836
Abstract
Introduction: Cholesteryl ester transfer protein (CETP) gene has been implicated in lipid metabolism. However, little is known about the impact of this gene on coronary heart disease (CHD) patients and its interaction with diet.
Hypothesis: To evaluate whether the chronic consumption of a Mediterranean diet enriched in olive oil, compared with a Low fat diet, interacts with the rs3764261 SNP at CETP locus in order to modify lipid metabolism among MetS patients from the CORDIOPREV clinical trial
Methods: Plasma lipid concentrations and rs3764261 genotypes were determined in 424 MetS subjects participating in the CORDIOPREV clinical trial. Gene-diet interactions were analyzed after a year of dietary intervention (Mediterranean diet (35% fat, 22% MUFA) vs Low fat
diet (28% fat, 12% MUFA))
Results: We found significant gene-diet interactions between rs3764261 SNP and the dietary pattern for HDL-C (P=0.006) and triglyceride concentrations (P=0.040). Specifically, after 12 months of Mediterranean diet intervention, subjects who were carriers of the minor T allele (TT+TG) displayed higher plasma HDL-C concentrations (P=0.021) and lower triglycerides (P=0.020) compared with homozygous for the major allele (GG). In contrast, in the Low fat intervention group no significant differences were found between CETP genotypes after 12 months of dietary treatment.
Conclusions: Our data support the notion that a chronic consumption of a Mediterranean diet may play a contributing role in triggering lipid metabolism by interacting with the rs3764261 SNP at CETP gene locus in MetS patients
Article Information
vol. 130 no. Suppl 2 A19836
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Antonio Garcia Rios;
- Francisco Gomez-Delgado;
- Ana Isabel Perez Caballero;
- Andreea Corina-Baba;
- Vanesa Navarro-Martos;
- Carmen Marin-Hinojosa;
- Jose Lopez-Miranda;
- Pablo Perez-Martinez
- Lipid and Atherosclerosis Unit, Lipid and Atherosclerosis Unit. IMIBIC/Reina Sofia Univ Hosp/Univ of Cordoba, Córdoba, Spain
Abstract 18939: Haplotype-Dependent Differential Regulation of the Human AT1R Gene is Exacerbated by Age: Effects on Tissue Inflammatory and Redox Milieu
Anita Rana, Sudhir Jain, Nitin Puri, Meenakshi Kaw, Ashok Kumar
Circulation. 2014;130:A18939
Abstract
Age-associated inflammation and redox imbalance underlie etiopathogenesis of cardiovascular-renal diseases, including hypertension and end organ damage. Angiotensin II (Ang II), via activation of the AT1R, contributes to the development and progression of these pathophysiologies. We have identified two haplotype blocks of single nucleotide polymorphisms (SNPs) in the hAT1R gene: haplotype II (Hap II: -810A, -713G, -214C, -153G) and I (Hap I: -810T, -713T, -214A, -153A). In clinical studies, Hap I is linked to human hypertension. This study examines haplotype-dependent and age-associated transcriptional regulation of the hAT1R gene. In this regard, we have engineered transgenic (TG) mice with either haplotype of the hAT1R gene using a 166-kb bacterial artificial chromosome. ChIP assay shows increased RNA-Pol II binding (~1.6 fold higher) to the chromatin extracts from renal tissues of adult (4-6 months) male Hap I-TG mice with increased hAT1R expression (~6 fold higher). This was accompanied by higher baseline blood pressure in Hap I-TG mice (Hap I- 129±3 vs. Hap II- 116±4, p18 months), male mice were used for this part of the study. hAT1R expression increases with age in both haplotypes; however, this increase is significantly higher in Hap I-TG mice (3.17±0.5 to 5.5±0.75 fold) as opposed to mice with Hap II (1.1±0.2 to 1.8±0.1 fold). Age-associated change ([[Unable to Display Character: ∆]]) in inflammatory and redox markers was significantly (p<0.05) greater in TG mice with Hap I including, IL1 (4.6±0.8 vs. 2.1±0.49 fold), IL6 (4.0±0.69 vs. 2.1±0.2 fold) and NOX1 (8.3±0.4 vs. 2.5±0.6 fold). This is accompanied by age-associated reduction in levels of antioxidant defenses (SOD1: 0.97±0.0 vs. 1.4±0.1 fold; HO1: 0.77±0.1 vs. 1.3±0.2 fold) and pro-survival genes including, NAMPTS (2.1 folds lower in Hap I vs. Hap II) and SIRT1 (1.8 fold lower in Hap I vs. Hap II). Thus, haplotype-dependent transcriptional regulation of the hAT1R gene causes increased hAT1R expression and blood pressure, in Hap I TG mice. Importantly, aging exacerbates this differential gene-expression regulation, further increasing hAT1R and promoting a prooxidant/inflammatory milieu in mice with Hap I.
Article Information
vol. 130 no. Suppl 2 A18939
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Anita Rana;
- Sudhir Jain;
- Nitin Puri;
- Meenakshi Kaw;
- Ashok Kumar
- Physiology and Pharmacology, The Univ of Toledo, Toledo, OH
Abstract 18728: Discovery Lipidomic and Metabolomic Profiling Predicts Longitudinal Changes in Metabolic Risk Factors
Christine M Willinger, Xiaoyan Yin, Peter Juhasz, Paul Courchesne, Pieter Muntendam, Neal Gordon, Aram Adourian, Martin G Larson, Daniel Levy
Circulation. 2014;130:A18728
Abstract
BACKGROUND: Metabolic syndrome is associated with increased risk for cardiovascular disease and type 2 diabetes, but the molecular mechanisms underpinning its constituent risk factors are unclear. We sought to identify predictive markers of metabolic risk by testing for associations of lipids and metabolites with longitudinal changes in metabolic traits.
METHODS: Discovery liquid chromatography-tandem mass spectrometry profiling of 154 lipids and gas chromatography-mass spectrometry profiling of 119 metabolites was conducted on plasma samples from 554 Framingham Heart Study participants with baseline and follow-up clinic examinations (5-7 years later) at which body mass index (BMI), triglycerides (TG), HDL cholesterol (HDL-C), and glucose were measured. Analytes were tested for association with longitudinal changes (Δ) in metabolic traits using general linear models, and multimarker panels were selected using forward selection.
RESULTS: Our single marker analyses revealed distinct signatures of longitudinal changes in metabolic risk factors. Markers associated with ΔBMI were 1-palmitoyl lysophosphatidic acid (LPA 16:0; p=2.8×10-4), its precursor lysophosphatidylcholine (LPC 16:0; p=6.7×10-5), and four other LPC species. Subclasses of sphingomyelins (SMs) were associated with changes in TG, HDL-C, and glucose. Sphingadiene (d18:2) variants were associated with ΔHDL-C [SM(d18:2/24:1), p=2.8×10-6; SM (d18:2/20:0), p=1.2×10-4]. Canonical SMs were associated with ΔTG [SM (d18:1/16:0), p=1.8×10-5; SM (d18:1/17:0), p=2.1×10-5]. Two dihydrosphingosine (d18:0) variants were associated with Δglucose [SM 36:0, p=5.0×10-5; SM (d18:0/24:0), p=3.2×10-4]. Metabolite markers for ΔTG included quinic acid (p=9.1×10-5) and sitosterol (p=1.0×10-4). Top markers were selected in multimarker panels that explained a significant proportion of longitudinal change in each metabolic trait (2.5-15.3%) beyond baseline covariates.
CONCLUSIONS: Using lipidomic and metabolomic profiling in parallel, we identified lipid-centric signatures of longitudinal changes in metabolic traits and demonstrated their predictive power. Our results suggest that specific derangements in lipid metabolic pathways may underlie metabolic risk.
Article Information
vol. 130 no. Suppl 2 A18728
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Christine M Willinger1;
- Xiaoyan Yin2;
- Peter Juhasz3;
- Paul Courchesne1;
- Pieter Muntendam4;
- Neal Gordon5;
- Aram Adourian6;
- Martin G Larson7;
- Daniel Levy1
- 1Framingham Heart Study, NHLBI, Framingham, MA
- 2Neurology, Boston Univ, Boston, MA
- 3Cntr of Mass Spectrometry and Discovery Proteomics, Biogen Idec, Cambridge, MA
- 4Leadership, scPharmaceuticals, LLC, Lexington, MA
- 5Rsch Projects, 121 Bio, LLC, Cambridge, MA
- 6Leadership, BG Medicine, Waltham, CT
- 7Biostatistics, Boston Univ, Boston, MA
Abstract 18405: Epidrug Treatment Rescues Proliferation and Differentiation in Human Cardiac Mesenchymal Cells of Type 2 Diabetic Patients: A Case of Epimetabolic Memory
Francesco Spallotta, Chiara Cencioni, Antonella Farsetti, Andreas M Zeiher, Carlo Gaetano
Circulation. 2014;130:A18405
Abstract
Introduction: The origin of metabolic/epigenetic memory remains elusive. It, however, may be at the basis of chronic diseases and functional alteration associated to ageing and cardiovascular disease.
HYPOTHESIS: Diabetes is known to alter cellular metabolism causing defects in tissue/organ regeneration. In this study we asked whether human cardiac stromal cells from diabetic and normoglycaemic voluntary donors revealed differences associated to their original metabolism and whether the alterations could be rescued by epigenetically active drugs.
METHODS and RESULTS: D-CMSC were characterized by a reduced proliferation rate, diminished phosphorylation at histone H3 serine 10 (H3S10P), decreased differentiation potential, and premature cellular senescence. A global histone code profiling of D-CMSC revealed that acetylation on histone H3 lysine 9 (H3K9Ac) and lysine 14 (H3K14Ac) was decreased, whereas the trimethylation of histone H3 lysine 9 and lysine 27 significantly increased. These observations were paralleled by a downregulation of the GCN5- related N-acetyltransferases (GNAT) p300/CBP-associated factor and its isoform 5-a general control of amino acid synthesis (GCN5a), determining a relative decreasein total HAT activity. DNA CpG island hypermethylation was detected at promoters of genes involved in cell growth control and genomic stability. Remarkably, treatment with the GNAT proactivator SPV106 restored normal levels of H3K9Ac and H3K14Ac, reduced DNA CpG hypermethylation, and recovered D-CMSC proliferation and differentiation.
CONCLUSIONS: This study investigated the diabetes-associated alterations present in cardiac mesenchymal cells (CMSC) obtained from normoglycemic (ND-CMSC) and type 2 diabetic patients (D-CMSC), identifying the histone acetylase (HAT) activator pentadecylidenemalonate 1b (SPV106) as a potential pharmacological intervention to restore cellular function. These results suggest that epigenetic interventions may reverse alterations in human CMSC obtained from diabetic patients.
Article Information
vol. 130 no. Suppl 2 A18405
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Dept. of Cardiology-Internal Medicine Clinic III, Goethe Univ, Frankfurt am Main, Germany
- 2Consiglio Nazionale delle Ricerche, Isitituto Neurobiologia e Biologia Cellulare, Roma, Italy
Abstract 16991: Multiple Peripheral Blood Protein Biomarkers Are Associated with Appropriate ICD Therapy in the DISCERN Study
James Wingrove, Phil Beineke, Karen Fitch, Dawood Darbar, Steven Rosenberg
Circulation. 2014;130:A16991
Abstract
Background: The decision to implant an ICD for primary prevention of ventricular arrhythmia is challenging and has relied on ventricular ejection fraction as a primary decision making factor, although improved risk stratification methodologies are clearly needed. Recent evidence suggests that inflammation plays a role in the generation of ventricular arrhythmias; we and others have demonstrated that increased C-reactive protein (CRP) levels are associated with appropriate ICD therapy. We have expanded these initial observations by interrogating a large set of circulating proteins in a population of patients with ICDs.
Methods: Levels of 103 proteins in plasma were assessed in a case:control cohort of 152 subjects (72 cases) enrolled in DISCERN (NCT00500708), a multi-center study designed to identify markers associated with ventricular arrhythmia. Assays were chosen from an existing set of proteins largely associated with inflammation. Patients had ejection fractions of <50%, 92% were male and 91% had ischemic HF. ICD firing data was captured for all subjects; cases had appropriate adjudicated ICD therapy in response to VT/VF, controls had no appropriate therapy within two years of ICD implant.
Results: In a logistic regression model adjusting for age, sex and WBC, the levels of 10 proteins (NTproBNP, CRP, SAA, MMP9, sMET, CCL2, Ang2, Resistin, TNFR1, E-Selectin) were significantly associated with appropriate ICD therapy (p < 0.05). In an exploratory analysis, grouping subjects by number of events (events = 0, 1, >1) resulted in even stronger associations, with an additional 11 proteins identified (p < 0.05). Multiple biological pathways were represented in the set of proteins, including inflammation, cell adhesion, and glucose regulation.
Conclusions: Multiple protein biomarkers were significantly associated with appropriate ICD therapy, showing stronger associations in patients with multiple events. The use of such markers may provide an approach to risk stratify heart failure patients for ICD implantation.
Article Information
vol. 130 no. Suppl 2 A16991
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Rsch, CardioDx, Inc, Palo Alto, CA
- 2Rsch, CardioDx, Inc, Redwood City, CA
- 3Div of Cardiovascular Medicine, Vanderbilt Univ Sch of Medicine, Nashville, TN
Abstract 16718: The Long Noncoding RNA H19 Controls Vascular Ageing and Inflammation
Patrick Hofmann, Katharina M Michalik, Anuradha Doddaballapur, Stefanie Dimmeler, Reinier Boon
Circulation. 2014;130:A16718
Abstract
Long noncoding RNAs (lncRNAs) are endogenously expressed noncoding RNAs with a length of more than 200 nucleotides, which can regulate gene expression through various mechanisms. Laminar shear stress and the expression of the flow-induced transcription factor Krüppel like factor 2 (KLF2) decrease with ageing. We hypothesize that protective factors like shear stress and detrimental factors like ageing and inflammation regulate the expression of lncRNAs that in turn regulate endothelial cell (EC) functions.
Using next generation sequencing, we identified differentially regulated lncRNAs in several tissues of young and aged mice. The most profoundly regulated lncRNA was H19. Microarray profiling of lung ECs from 2 and 18 months old mice revealed a downregulation of H19 with ageing (-4.1 ± 1.2 fold), qPCR analysis of the same tissue from Ku80+/- progeria mice showed similar effects (-1.7 ± 0.4 fold) compared to WT littermates. In the aortic intima of 20 months old C57BL/6J mice, H19 was also downregulated (-3.7 ± 0.6 fold; P<0.01). siRNA- and LNA GapmeR-mediated knockdown of H19 in human umbilical vein ECs (HUVECs) reduced proliferation and increased the mRNA level of the senescence marker p21 (+2.0 ± 0.2 fold; P<0.01).
In contrast, shear stress significantly upregulated H19 in HUVECs (+3.6 ± 0.6 fold; P<0.01), while it was not upregulated under shear stress after silencing of KLF2. Consistently, H19 expression can be induced by overexpression of KLF2 (+5.0 ± 0.2 fold; P<0.05). Furthermore, the inflammatory activity of ECs was increased after silencing of H19 (ICAM1 +1.4 ± 0.1 fold and VCAM1 +2.3 ± 0.6 fold). Similarly, inflammatory activation of ECs by TNFα also reduced H19 levels (-2.3 ± 0.8 fold). H19 is predominantly located in the cytoplasm of HUVECs and was not bound to silenced or activated histones, suggesting that it exhibits its function independent of epigenetic transcriptional control mechanisms.
In summary, H19 expression is controlled by the shear stress induced transcription factor KLF2 and is downregulated with ageing and inflammation. Functionally, H19 knockdown induces a senescent-like phenotype and increases the inflammatory activity of ECs. Together these results identify H19 as a lncRNA that controls pivotal endothelial functions.
Article Information
vol. 130 no. Suppl 2 A16718
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Patrick Hofmann;
- Katharina M Michalik;
- Anuradha Doddaballapur;
- Stefanie Dimmeler;
- Reinier Boon
- Institute of Cardiovascular Regeneration, Univ Hosp Frankfurt, Frankfurt am Main, Germany
Abstract 15265: Exome-wide Association Study of the Human Metabolome in a Community-based Cohort
Eugene Rhee, Qiong Yang, Xuan Liu, Jennifer Ho, Susan Cheng, Clary Clish, Daniel Levy, Vasan Ramachandran, Thomas Wang, Christopher O’Donnell, Robert Gerszten
Circulation. 2014;130:A15265
Abstract
Introduction: Because metabolites are hypothesized to play key roles as markers and effectors of cardio-metabolic diseases, recent studies have sought to annotate the genetic determinants of circulating metabolite levels. We hypothesize that the study of low frequency and rare variants of potential functional significance will refine our understanding of the genetic determinants of human metabolism.
Methods: We analyzed the association between 217 plasma metabolites and exome variants captured on the Illumina HumanExome Beadchip in 2,076 Framingham Heart Study (FHS) participants. Of the >240,000 variants on the exome array, we restricted our analysis to the subset of 92,633 variants that were polymorphic and nonsynonymous, nonsense, or located in a splice site. We performed association analyses that relate each single variant to each metabolite, as well as gene-based burden tests that evaluate the aggregate effects of all variants within a gene. Statistical analyses accounted for number of SNPs or genes (for burden tests).
Results: We identified a total of 15 gene-metabolite associations, nine from single variant analysis (gene: metabolite): CTH (Cystathionine); BPIFA2 (Betaine); OR1F1 (Creatinine); TCF19 (Glycerol); GMPS (Xanthosine); SIGLEC6 (Creatine); ANKUB1 (Carnosine); KIAA1751 (Serotonin); MAP1A (TAG 58:6). (Bonferroni-adjusted significance threshold of P< 5.4 x 10-7 for all). All but the ANKUB1findings are attributable to rare variants with MAF 0.01 or less. We identified six additional associations from burden tests, including three at established human disease loci: HAL (histidine); PAH (phenylalananine); UPB1(ureidopropionate); TG (indole propionate); CDH5 (SM 22:1); PHLDB1 (LPE 18:0) (Bonferroni-adjusted significance threshold of P < 3.9 x10-6 for all).
Conclusion: Our findings highlight genes with a direct biochemical relationship with the given metabolite as well as unanticipated genetic effectors of select metabolites. We show how an examination of variants across the spectrum of allele frequency is able to identify independent association signals at select loci as well as generate a more integrated view of genetic determinants of metabolites.
Article Information
vol. 130 no. Suppl 2 A15265
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Eugene Rhee1;
- Qiong Yang2;
- Xuan Liu3;
- Jennifer Ho4;
- Susan Cheng5;
- Clary Clish6;
- Daniel Levy7;
- Vasan Ramachandran8;
- Thomas Wang9;
- Christopher O’Donnell10;
- Robert Gerszten1
- 1Cardiology, Massachusetts General Hosp, Boston, MA
- 2Biostatistics, Boston Univ Sch of Medicine, Boston, MA
- 3The Framingham Heart Study, Boston Univ Sch of Medicine, Boston, MA
- 4Cardiovascular Medicine, Boston Univ Sch of Medicine, Boston, MA
- 5Cardiology, Brigham and Women’s Hosp, Boston, MA
- 6Cardiology, Broad Institute, Boston, MA
- 7Cntr for Population Studies/Framingham Heart Study, National Institute of Health, Framingham, MA
- 8Cardiology, Boston Univ Sch of Medicine, Boston, MA
- 9Cardiovascular Medicine, Vanderbilt Heart & Vascular Institute, Nashville, TN
- 10Cardiology, National Institute of Health, Framingham, MA
Abstract 14885: CXCL4 Aggravates Mortality and Left Ventricular Dilation Following Myocardial Infarction by Polarizing Macrophages to a Pro-inflammatory M1 Phenotype
Yonggang Ma, Andriy Yabluchanskiy, Ryan Clark, Presley L Cannon, Elizabeth R Flynn, Yu-Fang Jin, Merry L Lindsey
Circulation. 2014;130:A14885
Abstract
CXCL4 deletion attenuates atherosclerosis and mesenteric ischemia/reperfusion injury in mice. CXCL4 roles following myocardial infarction (MI) have not been evaluated. We hypothesized that CXCL4 would exacerbate post-MI wound healing and cardiac dysfunction. Male C57BL/6J mice (n=82) of 3-6 months old were subjected to permanent coronary artery ligation. CXCL4 gene expression significantly increased after MI, peaking at day 5 (p<0.05 vs day 0). To investigate whether CXCL4 exacerbates the inflammatory response, the mice were treated with 5, 25, or 50 μg/kg/day CXCL4 or saline at 24 hours post-MI through osmotic mini-pump as an overexpression strategy. Saline treated mice showed 47% survival at day 7 post-MI (7 out of 15), while CXCL4 treatment with all 3 doses dramatically reduced the survival rate to 10% (1 out of 10, p<0.05). Autopsy evaluation revealed that 50% (4 of 8) of saline treated mice died of cardiac rupture, while CXCL4 treatment (50 μg/kg/day) showed 89% rupture rate (8 of 9). The infarct area was similar between saline and CXCL4 treated surviving mice (53±2% for saline and 53±4% for CXCL4, p=0.95). Compared to the saline group, CXCL4 treated mice showed significantly higher end-systolic and end-diastolic volumes (both p<0.05), indicating that CXCL4 exacerbated left ventricular dilation. Out of 84 inflammatory genes measured, CXCL4 treatment resulted in the decreased expression of Ccl3 and Ltb and increased expression of Ccr4 and Cxcl11 (all p<0.05 compared to saline treatment), indicating very specific signaling targets. In vitro, CXCL4 (5 μg/mL, 4 hour stimulation) polarized macrophages to a pro-inflammatory M1 phenotype, evidenced by upregulation of M1 markers (Ccl3, Ccl5, Il1β, and Il6, all p<0.05), and by downregulation of M2 marker Cd206 (p<0.05). In conclusion, CXCL4 exacerbated mouse death and left ventricular dilation post-MI by polarizing macrophages towards a pro-inflammatory M1 subtype.
Article Information
vol. 130 no. Suppl 2 A14885
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Yonggang Ma1;
- Andriy Yabluchanskiy1;
- Ryan Clark1;
- Presley L Cannon1;
- Elizabeth R Flynn1;
- Yu-Fang Jin2;
- Merry L Lindsey1
- 1Physiology and Biophysics, Univ of Mississippi Med Cntr, Jackson, MS
- 2Electrical and Computer Engineering, The Univ of Texas at San Antonio, San Antonio, TX
Abstract 20256: Clinical Presentation, Management and Longitudinal Follow-Up of Neonatal and Infants With Coronary Artery Fistula – A Multicenter Study From the Coronary Artery Fistula Registry
Srinath T Gowda, Shelby Kutty, Asnes Jeremy, Lourdes Prieto, Zhi Chen, Masaru Miura, Takeda Atsuhito, Athar Qureshi
Circulation. 2014;130:A20256
Abstract
Background: Neonatal coronary artery fistula is rare, and data regarding its clinical presentation, treatment, and long term outcome are lacking.
Methods: Data in all neonates and infants who underwent evaluation in the catheterization lab for coronary artery fistula (CAF) from Jan 1995 – Jan 2014 were reviewed from the multicenter CAF registry. The CAF were classified as proximal and distal for analysis. Results: A total of 21 patients were included from 12 centers.
Results: The median age and BSA was 0.26 yrs and 0.26 m2, respectively. At presentation, heart failure (HF) symptoms were noted in 14 / 21 (61%) pts; 11 had proximal and 10 had distal CAF. The largest and narrowest mean calibers of CAF were 6.9 and 3.3mm. Eleven pts underwent transcatheter closure (TCC), 7 surgical closure (SC), and 3 had medical observation only.
Procedural success was 100%,; intentional partial closure was performed in 1 patient with large distal fistula. Follow-up with median 2.3 ( 0.3-18 ) years in 90% pts revealed coronary events in 3 patients; 1 pt with large distal RCA fistula had myocardial infarction at 9 days post SC with ventricular dysfunction requiring thrombolytics and there was recovery of ventricular function. In 2/9 pts, follow-up angiography showed asymptomatic coronary thrombosis in 2; 1 with large distal RCA fistula and SC had complete RCA occlusion and remodeling with thread like vessels in the RCA at 2.5 yrs follow-up angiography. One patient with proximal LCA to left atrium fistula with severely dilated coronary sinus had SC, angiography 4 years later showed complete LCX occlusion from thrombus extension and retrograde filling from collaterals.
Conclusions: Neonates and infants with CAF frequently present with HF symptoms. They are treated successfully by transcatheter or SC, however, large distal CAF and proximal fistula with severely dilated coronary sinus are at risk for acute and chronic coronary events. Therefore, long-term follow-up coronary anatomical and functional evaluation is imperative for its evaluation and management. Partial closure of the large distal CAF may prevent coronary events following closure.
Article Information
vol. 130 no. Suppl 2 A20256
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Srinath T Gowda1;
- Shelby Kutty2;
- Asnes Jeremy3;
- Lourdes Prieto4;
- Zhi Chen5;
- Masaru Miura6;
- Takeda Atsuhito7;
- Athar Qureshi8
- 1Pediatric Cardiology, Children’s Hopsital of Michigan, Detroit, MI
- 2Pediatric Cardiology, Children’s Hosp and Med center, Omaha, NE 68114, NE
- 3Pediatric Cardiology, Yale Sch of Medicine, New Haven, CT
- 4Pediatric Cardiology, Cleveland Clinic, Cleveland, OH
- 5Pediatric Cardiology, Hunan Provincial Children’s Hosp, Hunan, China
- 6Pediatric Cardiology, Tokyo Metropolitan Children’s hospital, Tokyo, Japan
- 7Pediatric Cardiology, Hokkaido Univ Hosp, Sapporo, Kiribati
- 8Pediatric Cardiology, Texas Children’s Hosp, Houston, TX
Abstract 19074: Relationship Between Hospital Procedure Volume and Complications Following Congenital Cardiac Catheterization: a Report from the IMPACT® Registry
Andrew C Glatz, Natalie M Jayaram, Michael L O’Byrne, Yan Li, Paul S Chan, Praneet K Sharma, Lisa Bergersen, John A Spertus
Circulation. 2014;130:A19074
Abstract
Background: The association between institutional procedural volume and outcomes has been demonstrated for many procedures within the field of cardiology, but whether this relationship pertains to congenital cardiac catheterization is unknown.
Methods: Using the IMPACT® (Improving Pediatric and Adult Congenital Treatment) Registry, we identified all catheterizations between 2011 and 2013 at 64 U.S. centers. Programs were categorized as small (<150 cases/year), medium (150 to 300 cases/year), large (300-500 cases/ year), and very large (>500 cases/ year). Hierarchical logistic regression, adjusted for patient and procedural characteristics, was used to determine the association between annual procedural volume and occurrence of a major adverse event (cardiac arrest; arrhythmia requiring permanent pacemaker; tamponade; air embolus; embolic stroke; device malposition, thrombus, or embolization; new requirement for dialysis; event requiring extracorporeal membrane oxygenation or left ventricular assist device; unplanned cardiac, vascular, or other surgery; or subsequent cardiac catheterization).
Results: Among 64 hospitals, there were 19 small, 15 medium, 19 large, and 11 very large programs. Among 33,825 catheterizations, a major adverse event occurred in 711 (2.1%). In unadjusted analysis, major adverse events differed based upon annual volume, with a major adverse event occurring in 79 (2.8%) cases at small, 145 (2.3%) cases at medium, 234 (2.1%) cases at large, and 253 (1.9%) cases at very-large programs (p=0.01). After adjusting for patient and procedural risk factors, there was no association between annual procedure volume and occurrence of a major adverse event (odds ratio 0.96 [95% Confidence Interval=0.90, 1.04] per increase in annual volume of 100, p=0.32). There was no interaction between procedure-type risk group and volume on outcomes (p=0.49).
Conclusions: Although the unadjusted risk of adverse events was lower in centers performing more catheterizations for congenital heart disease, this association between center volume and outcomes did not persist after adjustment for patient and procedural case-mix. The hospital sample size and low event rate may have limited our ability to detect significant differences.
Article Information
vol. 130 no. Suppl 2 A19074
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Andrew C Glatz1;
- Natalie M Jayaram2;
- Michael L O’Byrne3;
- Yan Li4;
- Paul S Chan5;
- Praneet K Sharma5;
- Lisa Bergersen6;
- John A Spertus5
- 1Div of Cardiology, The Children’s Hosp of Philadelphia, Philadelphia, PA
- 2Div of Cardiovascular Rsch, Saint Luke’s Mid America Heart Institute and Children’s Mercy Hosps and Clinics, Kansas City, MO
- 3Div of Cardiology, The Children’s Hosp of Philadelphia and The Univ of Pennsylvania Perelman Sch of Medicine, Philadelphia, PA
- 4Dept of Cardiovascular Rsch, Saint Luke’s Mid America Heart Institute, Kansas City, MO
- 5Div of Cardiology, Saint Luke’s Mid America Heart Institute, Kansas City, MO
- 6Div of Cardiology, Boston Children’s Hosp, Boston, MA
Abstract 17395: Health Related Quality of Life in Patients with Congenital Heart Disease: a Meta-Analysis
Peter C Kahr, Robert M Radke, Stefan Orwat, Helmut Baumgartner, Gerhard-Paul Diller
Circulation. 2014;130:A17395
Abstract
Background: With improving survival rates the focus of attention in congenital heart disease (CHD) has shifted from mortality to morbidity, functional capacity and quality of life (QoL). Due to the heterogeneity of methods and results from relatively small individual trials that have assessed QoL in the past, the impact of CHD complexity on QoL remains unclear.
Methods/Results: We performed a systematic review of studies that report primary data from interviews/questionnaires for QoL. Overall, 234 studies were included in the analysis. In total, results of more than 47,400 CHD patients (46% females) at an average age of 24 years were reported. Across all studies, more than 90 different questionnaires were utilized to evaluate different aspects of QoL, with 38% of studies combining ≥2 questionnaires in one study. The most commonly used questionnaire was the SF36 form (70 studies). When combining data on QoL from publications employing the SF36 using a ratio of the means method, we found that QoL in two domains was reduced in patients with moderate or complex CHD: Compared with healthy patients, CHD patients scored lower in Physical Functioning (0.96 [0.93-0.99] and 0.91 [0.88-0.95] for medium and high complexity;p<0.01) and General Health (0.95 [0.91;0.99] and 0.91 [0.86;0.95];p<0.01). No such effect was evident in those patients with simple cardiac lesions or in the other domains of the SF36 (see Figure).
Conclusions: Despite an exponentially increasing number of QoL studies in CHD, no standardized approach for measuring and reporting QoL has emerged and the published results are heterogeneous. In aggregation, however, our results suggest that QoL is impaired in CHD patients with moderate or complex CHD, while no such impact could be established in patients with simple defects. To improve comparability between studies, standardizing measurement of QoL in CHD is required and should be attempted.
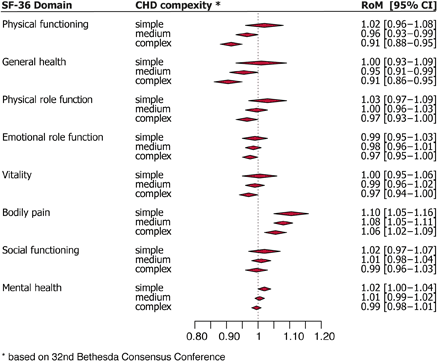
Article Information
vol. 130 no. Suppl 2 A17395
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Peter C Kahr;
- Robert M Radke;
- Stefan Orwat;
- Helmut Baumgartner;
- Gerhard-Paul Diller
- Div of Adult Congenital and Valvular Heart Disease, Univ Hosp Muenster, Muenster, Germany
Abstract 17071: Growth Asymmetry, Head Circumference and Neurodevelopmental Outcomes in Infants with Single Ventricles
Thomas A Miller, Victor Zak, Peter Shrader, Chitra Ravishankar, Victoria L Pemberton, Jane W Newburger, Amanda J Shillingford, Linda M Lambert, Renee Sananes, Marc E Richmond, James F Cnota, Daphne T Hsu, Stephen G Miller, Sinai C Zyblewski, Richard V Williams, Pediatric Heart Network Investigators
Circulation. 2014;130:A17071
Abstract
Poor somatic growth is common in infants with single ventricle (SV) physiology and has been linked to increased morbidity and impaired neurodevelopment. Asymmetry in somatic growth, a potential brain-sparing adaptation, is important in predicting outcomes in premature and small for gestational age (SGA) infants.
Objectives: To assess variability in growth asymmetry and its associations with neurodevelopment in infants with SV.
Methods: We analyzed growth asymmetry (weight for age z-score (WAZ) minus head circumference for age z-score (HCAZ)), relative head growth (change in cm/change in kg), HCAZ, and change in HCAZ from baseline to pre-Glenn in subjects prospectively enrolled in the Pediatric Heart Network Infant Single Ventricle (ISV) trial. Associations between these indices and results of the Psychomotor Developmental Index (PDI) and Mental Developmental Index (MDI) of the Bayley Scales of Infant Development-II (BSID) at 14 months were assessed.
Results: Of the 230 patients enrolled in ISV, complete biometric data and BSID results were available in 168 (73%). For this cohort, age at enrollment was 21±9 days, age at pre-Glenn was 167±52 days, gestational age was 38.3±1.4 weeks, and 71% were male. Growth asymmetry varied across the cohort at enrollment (0.43 ±1.02, range -2.85 to 4.84) and the pre-Glenn visit (-0.23 ±1.21, range -4.45 to 3.00) as did the relative head growth (2.40±0.86, range 0.50 to 8.00). BSID scores were not associated with indices of growth asymmetry. In univariate analysis, larger pre-Glenn HCAZ correlated with higher MDI (r=0.21, p=0.006) and PDI (r=0.38, p<0.001) and greater increase in HCAZ from enrollment to pre-Glenn was associated with higher PDI (r=0.15, p=0.049). In multivariable modeling adjusting for site, serious adverse events, stage 1 length of stay, and height at 14 months, pre-Glenn HCAZ was an independent predictor of PDI (p=0.03), but not MDI. For each one unit Z-score increase in pre-Glenn HCAZ, the predicted PDI score increased by 2.5 points.
Conclusions: In infants with SV, BSID scores were associated with pre-Glenn HCAZ but not with the degree of asymmetric growth. Future studies should explore why asymmetric growth that seems important in premature and SGA infants appears less relevant in infants with SV.
Article Information
vol. 130 no. Suppl 2 A17071
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Thomas A Miller1;
- Victor Zak2;
- Peter Shrader2;
- Chitra Ravishankar3;
- Victoria L Pemberton4;
- Jane W Newburger5;
- Amanda J Shillingford6;
- Linda M Lambert7;
- Renee Sananes8;
- Marc E Richmond9;
- James F Cnota10;
- Daphne T Hsu9;
- Stephen G Miller11;
- Sinai C Zyblewski12;
- Richard V Williams1,
- Pediatric Heart Network Investigators4
- 1Pediatrics, Univ of Utah, Salt Lake City, UT
- 2PHN, New England Rsch Institutes, Watertown, MA
- 3Pediatrics, Children’s Hosp of Philadelphia, Philadelphia, PA
- 4NHLBI, NIH, Bethesda, MD
- 5Cardiology, Children’s Hosp Boston, Boston, MA
- 6Pediatrics, Children’s Hosp of Wisconsin, Milwaukee, WI
- 7Surgery, Univ of Utah, Salt Lake City, UT
- 8Pediatrics, Hosp for Sick Children, Toronto, Canada
- 9Pediatrics, Columbia Univ Med Cntr, New York, NY
- 10Pediatrics, Cincinnati, Cincinnati, OH
- 11Pediatrics, Duke Univ Med Cntr, Durham, NC
- 12Pediatrics, Med Univ of South Carolina, Charleston, SC
Abstract 16992: The Impact of Pulmonary Insufficiency on Patients with Surgically Repaired Pulmonary Stenosis vs. Tetralogy of Fallot by Cardiac Magnetic Resonance
Michael R Joynt, Sunkyung Yu, Adam L Dorfman, Maryam Ghadimi Mahani, Prachi P Agarwal, Jimmy C Lu
Circulation. 2014;130:A16992
Abstract
Background: Patients with repaired pulmonary stenosis (PS) and tetralogy of Fallot (TOF) both develop pulmonary insufficiency (PI) leading to right ventricular (RV) dilatation and dysfunction. Cardiac magnetic resonance (CMR) plays a key role in determining timing of pulmonary valve replacement (PVR) in TOF, but it is unclear whether these criteria can be extrapolated to patients with PS. We aimed to compare the differential effect of pulmonary insufficiency on RV volume and systolic and diastolic function in patients with surgically repaired PS and TOF.
Methods: All patients with surgically repaired PS, undergoing CMR from 2007-2013, were matched 1:2 by age and pulmonary regurgitant fraction with TOF patients. Patients with prior PVR were excluded. Right and left ventricular (LV) ejection fraction (EF), end-diastolic volume (EDV), and presence of end-diastolic forward flow in the pulmonary artery (EDFF) were compared. Using feature tracking software (Tomtec, Unterschleissheim, Germany), RV longitudinal systolic strain and diastolic strain rate were measured from a 4-chamber slice, and RV circumferential strain from the most basal short-axis slice with circumferential RV myocardium.
Results: In 24 PS patients (mean 40.7 ± SD 13.3 years old, 41.7% male) with pulmonary regurgitant fraction of 41.5 ± 17.1% and 47 TOF patients (mean 39.1 ± SD 12.5 years old, 55.3% male) with pulmonary regurgitant fraction of 40.9 ± 16.3%, there was no difference in RV EDV (153.3 vs. 153.5 mL/m2, p = 0.99), presence of EDFF (83.3 vs. 61.7%, p = 0.10), or RV longitudinal diastolic strain rate (1.27 vs. 1.13, p = 0.13). However, PS patients had preserved RV EF (54.3 vs. 48.0%, p <0.0001) and LV EF (57.7 vs. 54.8%, p = 0.05) compared to TOF patients, predominantly due to difference in RV circumferential (-15.8 vs. -11.8, p <0.0001) rather than longitudinal strain (-18.0 vs. -15.9, p = 0.04).
Conclusions: With the same degree of PI, PS patients have similar RV dilatation and diastolic dysfunction compared to TOF patients. However, RV EF is preserved, predominantly due to differences in outflow tract function, as is LV EF. This may reflect different degrees of scarring and ventricular-ventricular interaction, and suggests CMR criteria for PVR may not be identical for patients with PS vs. TOF.
Article Information
vol. 130 no. Suppl 2 A16992
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Michael R Joynt1;
- Sunkyung Yu1;
- Adam L Dorfman1;
- Maryam Ghadimi Mahani2;
- Prachi P Agarwal3;
- Jimmy C Lu1
- 1Pediatric Cardiology, Univ of Michigan, Ann Arbor, MI
- 2Div of Cardiothoracic Radiology, Univ of Michigan, Ann Arbor, MI
- 3Radiology, Univ of Michigan, Ann Arbor, MI
Abstract 16297: High Quality Care is Associated With Lower Costs for Congenital Heart Surgery Across US Children’s Hospitals
Sara K Pasquali, Jeffrey P Jacobs, Edward L Bove, J. W Gaynor, Xia He, Michael G Gaies, Jennifer C Hirsch-Romano, John E Mayer, Eric D Peterson, Nelangi M Pinto, Samir S Shah, Matt Hall, Marshall L Jacobs
Circulation. 2014;130:A16297
Abstract
Introduction: There is currently a focus on both optimizing healthcare quality and reducing costs, or high “value” care. However, the care of children undergoing heart surgery requires significant investment of resources, and it is unclear whether it is possible to deliver high quality care, but also at a low cost. We evaluated the quality-cost relationship across a large multi-center cohort.
Methods: Clinical data from The Society of Thoracic Surgeons Database were linked to cost data from the Pediatric Health Information Systems Database for children 0-18 yrs undergoing heart surgery at hospitals participating in both datasets (2006-10). Hospital costs were modeled using Bayesian hierarchical methods, adjusting for procedural case mix [using Society of Thoracic Surgeons-European Association for Cardiothoracic Surgery (STAT) methods] and important patient characteristics. Adjusted mortality rates were compared across hospital cost tertiles. Complications and length of stay (LOS) were also examined.
Results: Overall, 30,670 patients (27 hospitals) were included. Median adjusted cost/case was $82,360 and varied 5-fold across hospitals. Hospitals in the lowest cost tertile tended to be larger volume centers treating higher complexity patients (26.5% vs. 22.8% in STAT category 4/5 in low vs. high cost centers, p<0.0001). Low cost hospitals had significantly lower adjusted mortality rates compared with the middle and high cost tertiles (2.5% vs. 3.8% and 3.5% respectively, p<0.0001, Figure). Lower cost hospitals also had shorter LOS and fewer major complications.
Conclusions: In this analysis, the lowest cost hospitals also appeared to deliver the highest quality care for children undergoing heart surgery, despite treating a more complex patient population. These results suggest that initiatives aiming to improve outcomes, whether through quality improvement or regionalizing care, may also have the potential to lower costs.
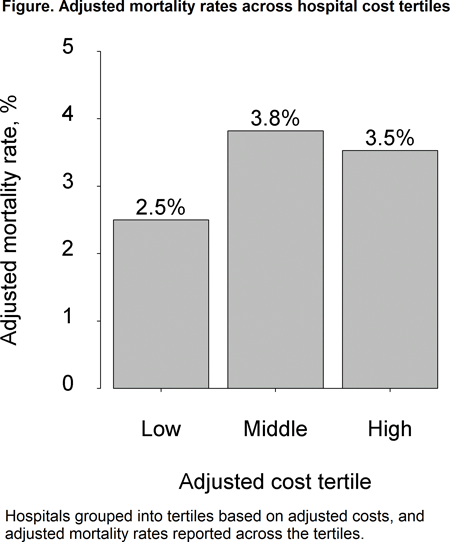
Article Information
vol. 130 no. Suppl 2 A16297
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Sara K Pasquali1;
- Jeffrey P Jacobs2;
- Edward L Bove3;
- J. W Gaynor4;
- Xia He5;
- Michael G Gaies1;
- Jennifer C Hirsch-Romano3;
- John E Mayer6;
- Eric D Peterson7;
- Nelangi M Pinto8;
- Samir S Shah9;
- Matt Hall10;
- Marshall L Jacobs11
- 1Pediatrics, Univ of Michigan, Ann Arbor, MI
- 2Cardiac Surgery, All Children’s Hosp, Saint Petersburg, FL
- 3Cardiac Surgery, Univ of Michigan, Ann Arbor, MI
- 4Surgery, Children’s Hosp of Philadelphia, Philadelphia, PA
- 5Statistics, Duke Univ, Durham, NC
- 6Surgery, Children’s Hosp Boston, Boston, MA
- 7Medicine, Duke Univ, Durham, NC
- 8Pediatrics, Primary Children’s, Salt Lake City, UT
- 9Pediatrics, Cincinnati Children’s Hosp Med Cntr, Cincinnati, OH
- 10Statistics, Children’s Hosp Association, Overland Park, KS
- 11Cardiac Surgery, Johns Hopkins, Baltimore, MD
Abstract 16381: Critical Care Nursing Experience and Education are Associated With Outcomes Following Pediatric Cardiac Surgery: An Analysis of the STS Congenital Heart Surgery Database
Patricia A Hickey, Sara K Pasquali, J. W Gaynor, Xia He, Kevin D Hill, Jean A Connor, Kimberlee Gauvreau, Marshall L Jacobs, Jeffrey P Jacobs, Jennifer C Hirsch-Romano
Circulation. 2014;130:A16381
Abstract
Background: Little is known about the relationship between nursing characteristics and pediatric cardiac surgery outcomes. We evaluated the association of critical care nursing education and experience with outcomes in a large multi-center cohort of children undergoing cardiac surgery.
Methods: Data from Children’s Hospital Association members participating in a nursing survey were linked to clinical data from the STS Congenital Database for children (0-18 yrs) undergoing cardiac surgery (2010-11). Level of nursing education and years of clinical experience were assessed for nurses in the unit caring for post-operative cardiac patients. Association of these variables with outcome was examined using multivariable logistic regression models accounting for within-center clustering and differences in case mix and patient characteristics across hospitals. Outcomes included in-hospital mortality, complication rate, and failure-to-rescue rate (FTR; mortality rate in those with a complication).
Results: Overall, 15,463 patients (29 hospitals) were included. The in-hospital mortality rate was 2.8%, post-operative complications occurred in 42.4%, and the FTR rate was 6.4%. In adjusted analysis, units with a higher proportion of nurses with a Bachelor of Science in nursing (BSN) education had lower complication rates (OR for 10% increase = 0.85, 95% CI 0.76-0.96, p<0.01). Units with a higher proportion of nurses with >2 years of experience had lower mortality rates (OR for 10% increase = 0.91, 95% CI 0.84-0.99, p=0.025). Results were similar when center volume was added to the models, suggesting that the relationship of these nursing characteristics with outcome is independent of volume. Neither nursing variable was associated with FTR in our analysis.
Conclusions: This study demonstrates that higher levels of critical care nursing education are associated with fewer complications following pediatric cardiac surgery, and confirms the results of previous analyses demonstrating the association of nursing education and experience with lower mortality. These data may help to inform decisions about nursing resource allocation and retention strategies to optimize outcomes for this complex population.
Article Information
vol. 130 no. Suppl 2 A16381
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Patricia A Hickey1;
- Sara K Pasquali2;
- J. W Gaynor3;
- Xia He4;
- Kevin D Hill5;
- Jean A Connor1;
- Kimberlee Gauvreau1;
- Marshall L Jacobs6;
- Jeffrey P Jacobs7;
- Jennifer C Hirsch-Romano8
- 1Pediatrics, Children’s Hosp Boston, Boston, MA
- 2Pediatrics, Univ of Michigan, Ann Arbor, MI
- 3Surgery, Children’s Hosp of Philadelphia, Philadelphia, PA
- 4Statistics, Duke Univ, Durham, NC
- 5Pediatrics, Duke Univ, Durham, NC
- 6Surgery, Johns Hopkins, Baltimore, MD
- 7Surgery, All Children’s Hosp, Saint Petersburg, FL
- 8Cardiac Surgery, Univ of Michigan, Ann Arbor, MI
Abstract 16190: National Trends in RSV Hospitalizations in Children With Hemodynamically Significant Heart Disease, 1997-2009
Patricia Y Chu, Christoph P Hornik, Jennifer S Li, Michael J Campbell, Kevin D Hill
Circulation. 2014;130:A16190
Abstract
Background: Children with hemodynamically significant heart disease (HS-HD) are at risk for morbidities and mortality due to respiratory syncytial virus (RSV). Palivizumab was approved for RSV prophylaxis in 1998. Guidelines released in December 2003 recommend palivizumab for all children < 2 yrs with HS-HD. We sought to define the impact of RSV prophylaxis in children with HS-HD by evaluating trends in U.S. RSV hospitalizations.
Methods: The 1997, ’00, ’03, ’06 and ’09 Healthcare Cost and Utilization Project (HCUP) Kids’ Inpatient Databases (KID) were used to estimate U.S. RSV hospitalizations in children < 2 yrs, overall and in those with HS-HD, using standard HCUP weighting methods. RSV was defined by ICD-9-CM codes for RSV infection. HS-HD was defined using ICD-9-CM codes from the Clinical Classifications Software for congestive heart failure, or an ICD-9-CM code for pulmonary hypertension, common truncus, common ventricle, or hypoplastic left heart syndrome.
Results: Our cohort included an estimated 461,491 RSV hospitalizations; 2,132 in children with HS-HD. Figure 1 depicts hospitalizations over time. There was no evident trend in number of overall RSV hospitalizations, however RSV hospitalizations in children with HS-HD declined by 39% from ’97 to ‘09. The largest decline was from ’97-’03. RSV hospitalizations in children with HS-HD relative to overall hospitalizations in children with HS-HD declined annually from ’97-’06 with a small increase in ‘09 (3.8%, 3.5%, 3.0%, 2.3% and 2.6% for successive analytic years). In 2009 mean hospital length of stay for children with HS-HD and RSV was 22.5 ± 2.1 days.
Conclusions: RSV disease burden in children with HS-HD has declined since palivizumab approval. Much of this decline occurred before palivizumab was recommended for use in HS-HD, perhaps reflecting early adoption of prophylaxis, or greater awareness of alternative preventative strategies. RSV remains a significant cause of morbidity in children with HS-HD.
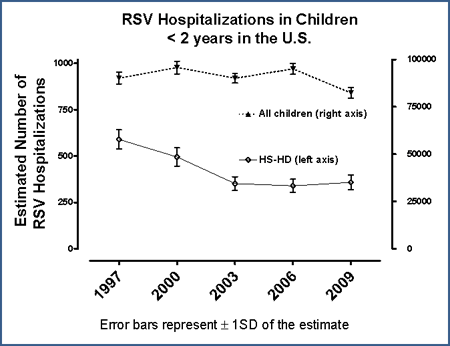
Article Information
vol. 130 no. Suppl 2 A16190
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Pediatrics, Duke Clinical Rsch Institute, Duke Univ Sch of Medicine, Durham, NC
- 2Pediatrics, Duke Clinical Rsch Institute, Duke Univ Med Cntr, Durham, NC
- 3Pediatric Cardiology, Duke Univ Med Cntr, Durham, NC
Abstract 14044: Prevalence of Rheumatic Heart Disaese in Ugandan School-Aged Population: Implications for Sub-Saharan Africa
Jacqueline Weinberg, Andrea Beaton, Jimmy C Lu, Twalib Aliku, Peter Dean, Lasya Gaur, Justin Godown, Peter Lwabi, Grace Mirembe, Emmy Okello, Alison Reese, Ashley Shrestha Astudillo, Janet Scheel, Catherine Webb, Gregory Ensing, Craig Sable
Circulation. 2014;130:A14044
Abstract
Background: The global prevalence of rheumatic heart disease (RHD) is estimated between 15.6 and 30 million based on population-based data, including 1 million sub-Saharan African school-aged children. A paucity of data from sub-Saharan Africa likely contributes to a significant underestimate of the true global burden of RHD. The 2012 World Heart Federation (WHF) echocardiographic guidelines allow for standardized assessment of RHD prevalence. We provide an updated estimate of RHD prevalence based on a large screening effort in northern Uganda.
Methods: Over a 1 week period, children aged 5-16 years at 5 local schools in Gulu, Uganda underwent echocardiography using the 2012 WHF guidelines. Gulu is the third largest city in Uganda with a population of 154,000. The results of this study were extrapolated to 51 other sub-Saharan African countries based on country-by-country population and age distribution. Morocco, Algeria, Tunisia, Libya, Egypt and South Africa were excluded. The prevalence of RHD in sub-Saharan African children of the same age was assumed to be similar to Gulu.
Results: Of 4,773 children screened, 4,561 studies were normal (95.6%), 52 met criteria for definite RHD (1.1%), and 140 met criteria for borderline RHD (2.9%). Of Uganda’s 35.4 million population, 12.1 million are between 5 and 16 years old, resulting in an estimate of definite and borderline RHD in Uganda of 132,704 and 349,856 school-aged children respectively. The total population in the 51 countries included is 852.5 million, with 27.2 million between 5 and 16 years old, resulting in an estimate of definite and borderline RHD in sub-Saharan Africa of 3.0 million and 7.9 million school-aged children respectively.
Conclusion: With systematic echocardiographic screening using 2012 WHF criteria, the estimated number of sub-Saharan African school-aged children with definite RHD is dramatically higher than prior reports, and borderline RHD cases may represent additional burden of disease. This may still be an underestimate, as it excludes children not well enough to attend school. Additionally the prevalance in older children and young adults is likely even higher. More systematic prevalence studies are needed to determine the true global burden of RHD.
Article Information
vol. 130 no. Suppl 2 A14044
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Jacqueline Weinberg1;
- Andrea Beaton1;
- Jimmy C Lu2;
- Twalib Aliku3;
- Peter Dean1;
- Lasya Gaur1;
- Justin Godown2;
- Peter Lwabi4;
- Grace Mirembe5;
- Emmy Okello4;
- Alison Reese1;
- Ashley Shrestha Astudillo2;
- Janet Scheel1;
- Catherine Webb2;
- Gregory Ensing2;
- Craig Sable1
- 1Div of Cardiology, Children’s National Health System, Washington, DC
- 2Div of Pediatric Cardiology, The Univ of Michigan, Ann Arbor, MI
- 3Dept of Paediatrics and Child Health, Gulu Univ, Gulu, Uganda
- 4Uganda Heart Institute, Uganda Heart Institute, Kampala, Uganda
- 5The Joint Clinical Rsch Cntr, The Joint Clinical Rsch Cntr, Kampala, Uganda
Abstract 12621: Factors Affecting Fontan Length of Stay: Results From the Single Ventricle Reconstruction Trial
Chitra Ravishankar, Eric Gerstenberger, Andrew M Atz, Jeremy Affolter, Timothy Bradley, Shaji Menon, Kurt Schumacher, Lynn A Sleeper, Carolyn Dunbar-Masterson, J W Gaynor, Bryan Goldstein, Heather Henderson, Jeff Jacobs, Alan B Lewis, Victoria L Pemberton, Christopher Petit, Nancy A Pike, Christian Pizarro, Ismee A Williams, Jane W Newburger
Circulation. 2014;130:A12621
Abstract
Background: In the Single Ventricle Reconstruction trial, subjects with hypoplastic left heart syndrome (HLHS) who received a right ventricle-to-pulmonary artery shunt (RVPAS) vs. modified Blalock-Taussig shunt (MBTS) had lower early postoperative mortality but more complications and worse RV ejection fraction at 14 mo. We explored the effect of shunt type and other patient and medical factors on postoperative length of stay (LOS) after the Fontan procedure.
Methods: Fontan postoperative course was ascertained from medical record review. Cox proportional hazards modeling was used to identify factors associated with LOS.
Results: Of 325 Fontan subjects, 323 were analyzed (1 death, 1 unknown discharge date). Of these, 285 (88%) had HLHS. Median age and weight at Fontan were 2.85 yr (IQR: 2.27, 3.37) and 12.7 kg (IQR: 11.4, 14.1), respectively. Fontan type was extracardiac in 178 (55%) and lateral tunnel in 145 (45%); 280 (87%) were fenestrated. Complications before discharge included pleural drainage >7 days in 58 (18%), arrhythmias in 50 (15%) and readmission within 30 days in 31 (10%). The RVPAS vs. MBTS subjects had similar LOS (median 11 d [IQR: 9, 18] vs. 10 d [IQR: 9, 13], P=.23). Independent risk factors for longer LOS were center (P<.01), LOS at Stage 2 (HR 1.02 for each additional day, P<.01), complications from birth to Fontan (HR 1.03 for each additional complication, P=.04). Use of circulatory arrest at Fontan (HR 0.61, P=.02) was associated with shorter LOS. When center was excluded from the multivariable model, pre-Fontan complications and use of circulatory arrest were no longer significant, and older age at Stage 2 (HR 1.08, P=.01) entered the model, predicting longer LOS. In 254 subjects with pre-Fontan Core Lab echocardiogram, ≥ moderate tricuspid regurgitation was independently associated with longer LOS both with center (HR 1.72, P<.01) and without center in the model (HR 1.49, P=.02). No variables from pre-Fontan catheterization (n=260) were associated with LOS.
Conclusion: In this multicenter prospective cohort, Norwood shunt type was not associated with Fontan LOS. Rather, global measures of earlier medical complexity including LOS at Stage 2 portend longer LOS after the Fontan procedure.
Article Information
vol. 130 no. Suppl 2 A12621
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Chitra Ravishankar1;
- Eric Gerstenberger2;
- Andrew M Atz3;
- Jeremy Affolter4;
- Timothy Bradley5;
- Shaji Menon6;
- Kurt Schumacher7;
- Lynn A Sleeper2;
- Carolyn Dunbar-Masterson8;
- J W Gaynor9;
- Bryan Goldstein10;
- Heather Henderson11;
- Jeff Jacobs12;
- Alan B Lewis13;
- Victoria L Pemberton14;
- Christopher Petit15;
- Nancy A Pike16;
- Christian Pizarro17;
- Ismee A Williams18;
- Jane W Newburger8,
- for the Pediatric Heart Network
- 1Pediatrics, The Children’s Hosp of Philadelphia, Philadelphia, PA
- 2Clinical and Statistical Science, New England Rsch Institute, Watertown, MA
- 3Pediatrics, Med Univ of South Carolina, Charleston, SC
- 4Critical Care Medicine, Children’s Hosp of Wisconsin, Milwaukee, WI
- 5Pediatrics, Hosp for Sick Children, Toronto, Canada
- 6Pediatrics, Primary Children’s Med Cntr, Salt Lake City, UT
- 7Pediatrics, Univ of Michigan Med Cntr, Ann Arbor, MI
- 8Cardiology, The Children’s Hosp of Boston, Boston, MA
- 9Cardiothoracic Surgery, Children’s Hosp of Philadelphia, Philadelphia, PA
- 10Pediatrics, The Children’s Hosp of Cincinnati, Cincinnati, OH
- 11Pediatrics, Duke Med Cntr, Durham, NC
- 12Cardiothoracic Surgery, Congenital Heart Institute of Florida, St Petersburg, FL
- 13Pediatrics, Children’s Hosp of Los Angeles, Los Angeles, AB
- 14NHLBI, NHLBI/NIH, Bethesda, MD
- 15Pediatrics, Sibley Heart Cntr, Atlanta, GA
- 16Pediatrics, UCLA, Los Angeles, CA
- 17Cardiothoracic Surgery, AI DuPont Hosp for Children, Wilmington, DE
- 18Pediatrics, Columbia Univ Med Cntr, NY, NY
Abstract 12447: Impact of 2007 American Heart Association Infective Endocarditis Prevention Guidelines on Endocarditis Related Pediatric Hospital Admissions: A U.S. Population-Based Study
Arpan R Doshi, Chandra Srinivasan, Hem H Desai, Heli P Bhatt, Mousumi Moulik
Circulation. 2014;130:A12447
Abstract
Purpose: The 2007 American Heart Association (AHA) guidelines for the prevention of infective endocarditis (IE) called for a major change in indications for antibiotic prophylaxis. Purpose of this study was to evaluate the national trends and mortality in IE; its associations with congenital heart disease (CHD); and the impact of 2007 AHA prevention guidelines.
Methods: This retrospective study examined inpatient admissions (age <20 yrs) for IE using nationally representative Kids’ inpatient database (KID), for the years 2006 and 2009. Complex sample analysis was performed to obtain weighted estimates of incidence and mortality rates on CHD associated with IE admissions. Categorized variables related to hospital and patient demographics were also studied.
Results: Out of 7.56 and 7.37 million total admissions in the years 2006 and 2009, there were 1,515 (95% CI 1,329-1,701) and 1,323 (95% CI 1,157-1,489) hospital admissions for IE, respectively (P = 0.072). While the incidence of IE-related admissions associated with isolated ASD increased significantly in year 2009 (P < 0.001), there was a decline (P = 0.020) in those admissions associated with isolated VSD. There was no significant change seen in the distribution of other CHD groups.
There was no significant change in overall mortality rate associated with IE-related admissions between the years 2006 and 2009 (P = 0.440). Non-white race and CHD were significant factors associated with mortality in IE-related admissions. Among CHD associated with IE admissions, isolated PDA, LVOTO, RVOTO were significantly associated with increased mortality on univariate analysis.
Conclusion: The 2007 AHA endocarditis prevention guidelines did not have a significant impact on the overall rate of hospital admissions for IE in calendar year 2009 compared to 2006. However, there was a significant decline in IE admissions with associated diagnosis of isolated VSD. The increase noted in the small number of IE admissions with associated isolated ASD is likely a confounding bias. Non-white race and CHD, particularly isolated PDA, LVOTO and RVOTO lesions were significantly associated with increased mortality in IE admissions on univariate analysis.
Article Information
vol. 130 no. Suppl 2 A12447
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Pediatric Cardiology, Univ of Texas Med Sch at Houston, Houston, TX
- 2Sch of Public Health, Univ of Texas, Houston, TX
- 3Sch of Public Health, Univ of Texas Med, Houston, TX
Abstract 11013: Resource Utilization for Non-Cardiac Hospital Admissions in Patients With Single Ventricle Congenital Heart Disease
Ian D Thomas, Michael D Seckeler
Circulation. 2014;130:A11013
Abstract
Background: Patients with single ventricle (SV) congenital heart disease (CHD) incur high hospital costs during staged surgical palliation. However, resource utilization for non-cardiac admissions in SV patients has not been reported. This study sought to compare costs for common non-cardiac hospital admissions between SV patients and patients without CHD.
Methods: A retrospective review of hospital discharge data from the University HealthSystem Consortium (UHC) from January 2011 through December 2013 was performed. UHC collects discharge data from 120 academic institutions and 302 affiliated hospitals. The database was queried for patients <18 years of age with ICD-9 codes for SV lesions: hypoplastic left heart syndrome (746.7), tricuspid atresia (746.1) or common ventricle (745.3). Neonates (<30 days old) were excluded to eliminate hospitalizations for Stage 1 surgical palliation. Primary diagnosis, direct cost, length of stay (LOS), ICU admission rate and mortality data were obtained. The eight most common non-cardiac admission diagnoses were compared between SV patients and non-CHD patients using t-test and Fisher’s exact test, as appropriate.
Results: The non-cardiac admission diagnoses, with ICD-9 codes, and comparisons between SV and non-CHD patients are shown in the Table. Total direct cost, LOS and ICU admission rate were higher for SV patients for all diagnoses with the exception of LOS for failure to thrive, which was not different between groups. Notably, hospital mortality was markedly higher for SV patients admitted for RSV bronchiolitis or pneumonia.
Conclusions: Hospital costs for common non-cardiac diagnoses are higher for patients with SV CHD. As long-term survival of SV CHD patients increase they will utilize a disproportionate amount of medical dollars, as our study shows. Further characterization of SV CHD patient costs will be important so steps can be taken to reduce or prevent hospitalization in these patients.
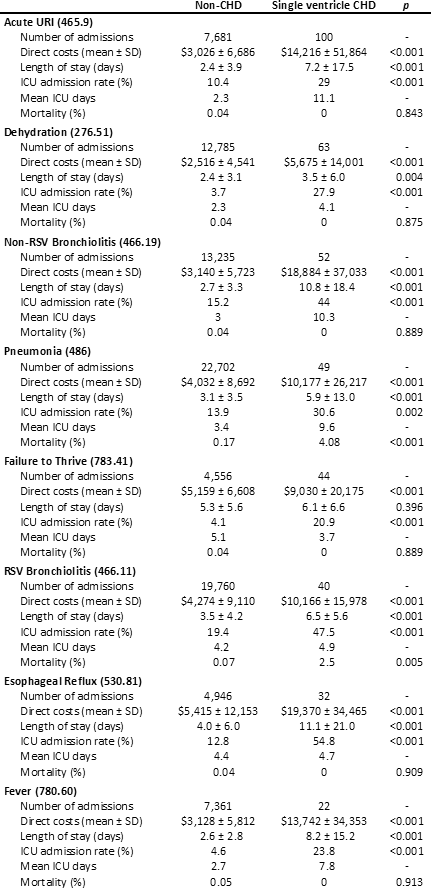
Article Information
vol. 130 no. Suppl 2 A11013
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Ian D Thomas;
- Michael D Seckeler
- Pediatrics, Univ of Arizona, Tucson, AZ
Abstract 11024: Quality of Life in Adolescents and Young Adults After the Fontan Operation: The Pediatric Heart Network Fontan Follow-Up Study
Karen Uzark, Victor Zak, Peter Shrader, Brian W McCrindle, Elizabeth Radojewski, James W Varni, Jill Handisides, Kaitlyn Daniels, Kevin Hill, Linda Lambert, Renee Margossian, Victoria L Pemberton, Wyman W Lai, Andrew M Atz
Circulation. 2014;130:A11024
Abstract
Objective: While life expectancy for children with single ventricle has improved, the potential impact of the Fontan circulation on overall quality of life (QOL) and functioning during transition to adulthood is largely unknown. We sought to describe QOL in a large multicenter cohort of adolescent and young adult Fontan survivors.
Methods: The Pediatric Quality of Life Inventory (PedsQL) was administered to 408 Fontan subjects ages 13-25 years enrolled in the Pediatric Heart Network Fontan Follow-up Study. Data were compared to control subjects without a chronic health condition from the PedsQL database (n=342) who were similar in age, gender, and race/ethnicity. Higher scores indicate better QOL. Scores > 1 standard deviation below the population mean reflect at-risk status for impaired QOL. Subjects also completed either the Child Health Questionnaire (CHQ-87, age< 19), or the Short Form 36 (SF-36, age ≥ 19).
Results: Mean age was 18.5 ± 3.4 years, 58% male, mean time post-Fontan 14.7 ± 3.8 years. Mean PedsQL scores for Fontan subjects were significantly lower than those for the control group for physical and psychosocial QOL, including emotional, social, and school/work QOL, all p<0.001. Overall, 45% of Fontan subjects had scores in the impaired range for physical QOL with 30% in the impaired range for psychosocial QOL. For each 1 year increase in age, the physical functioning score decreased by an average of 0.76 points (p=0.004) and the emotional functioning score decreased by an average of 0.64 points (p=0.03); among subjects ≥ 19 years of age, the physical functioning score decreased by an average of 2 points for each year increase in age (p=0.02). Physical and psychosocial functioning scores were not associated with Fontan type. Males reported higher scores than females for physical (p<0.001) and emotional QOL (p=0.002). PedsQL scale scores were significantly correlated with conceptually related CHQ-87 (p<0.001) and SF-36 scores, p<0.001.
Conclusion: Physical and psychosocial QOL are impaired in a substantial number of Fontan survivors and may decline with advancing age. Routine assessment of health-related QOL is essential to inform interventions to improve outcomes. The PedsQL allowed QOL assessment from pediatrics to young adulthood.
Article Information
vol. 130 no. Suppl 2 A11024
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Karen Uzark1;
- Victor Zak2;
- Peter Shrader2;
- Brian W McCrindle3;
- Elizabeth Radojewski4;
- James W Varni5;
- Jill Handisides6;
- Kaitlyn Daniels7;
- Kevin Hill8;
- Linda Lambert9;
- Renee Margossian10;
- Victoria L Pemberton11;
- Wyman W Lai12;
- Andrew M Atz13,
- for the Pediatric Heart Network Investigators
- 1Pediatrics, Univ of Michigan Congenital Heart Cntr, Ann Arbor, MI
- 2Statistics, New England Rsch Institutes, Inc., Watertown, MA
- 3Pediatrics, Univ of Toronto Hosp for Sick Children, Toronto, Canada
- 4Pediatrics Cardiology, The Hosp for Sick Children, Toronto, Canada
- 5Architecture, Texas A&M Univ, College Station, TX
- 6Pediatrics Nursing, Boston Children’s Hosp, Boston, MA
- 7Pediatrics Nursing, Children’s Hosp of Philadelphia, Philadelphia, PA
- 8Pediatrics, Duke Univ Med Cntr, Durham, NC
- 9Pediatric Cardiothoracic Surgery, Primary Children’s Hosp, Salt Lake City, UT
- 10Cardiology, Boston Children’s Hosp, Boston, MA
- 11Clinical Trials Specialist, National Heart, Lung, and Blood Institute, Bethesda, MD
- 12Pediatrics, Columbia Univ Med Cntr, New York, NY
- 13Pediatrics, Med Univ of South Carolina, Charleston, SC
Abstract 11174: A Novel Approach to Critical Congenital Heart Disease (CCHD) Screening at Moderate Altitude
Jason T Wright, Duster Mark, Leilani B Russell, Marci K Sontag, Cindy Eller, Christopher M Rausch
Circulation. 2014;130:A11174
Abstract
Introduction: Prenatal studies and postnatal physical exam leave 13- 55% of neonates with CCHD undiagnosed leading to presentation in extremis or death. The AHA has endorsed newborn pulse oximetry screening to capture these infants prior to hospital discharge. Moderate altitude can impart higher screening failure rates. We therefore evaluated a modified CCHD screening protocol in an attempt to reduce false positive screenings at a moderate altitude of 6200 feet (1890 m).
Methods: We prospectively enlisted well newborn infants greater than 35 weeks. Near 24 hours of life, trained nursing staff performed pulse oximetry on the right hand and either foot. Those with saturations ≤95% with <3% difference between hand and foot measurements passed. Infants with saturations <86% failed. Infants with saturations between 86-94% or >3% difference in saturations were placed in an oxygen hood with FiO2 designed to replicate sea level atmospheric oxygen tension for 20 minutes to accelerate neonatal transition. These infants were tested again up to 2 additional screens in room air with standard sea level protocol. Providers were notified and echocardiograms ordered for all infants deemed to have failed.
Results: A total of 2005 infants completed the protocol. The failure rate was 0.3% (7/2005), which was not different from the sea level rate of 0.2%. Sea level CCHD screening criteria would have given a failure rate of 0.75%. An additional 2.1% (42/2005) had incomplete screening and were not passing at the time the test was stopped. We found 5/7 (71.4%) infants failed secondary to low saturations, 1/7 (14.3%) failed secondary differential saturations, and 1 infant failed for multiple reasons. Three of the seven infants with failing screens were discharged prior to echocardiogram. None of the infants receiving echocardiograms had critical congenital heart disease.
Conclusions: We found a failure rate of 0.3% using an alternate algorithm adjusted for altitude. This failure rate approximates the overall screening failures reported at sea level and is significantly lower than prior reports at altitude. Additional research is needed specifically addressing sensitivity and positive predictive value for screening at moderate altitudes.
Article Information
vol. 130 no. Suppl 2 A11174
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Jason T Wright1;
- Duster Mark2;
- Leilani B Russell3;
- Marci K Sontag4;
- Cindy Eller5;
- Christopher M Rausch6
- 1Neonatology, Univ of Colorado, Aurora, CO
- 2Women’s Pavilion, Mother Baby Units, Univ of Colorado, Colorado Springs, CO
- 3Colorado Sch of Public Health Dept of Epidemiology, Univ of Colorado, Aurora, CO
- 4Dept of Epidemiology, Univ of Colorado Sch of Public Health, Aurora, CO
- 5Women’s Pavilion, Mother Baby Units Memorial Hosp part of Univ of Colorado Health, Univ of Colorado, Colorado Springs, CO
- 6Pediatric Cardiology, Univ of Colorado, Aurora, CO
Abstract 9473: Quantifying the Impact of Early Term Gestation Delivery Upon Resource Demands Following Neonatal Congenital Heart Surgery
Andrew H Smith, David A Parra, Bret A Mettler, David P Bichell, Ann Kavanaugh-McHugh
Circulation. 2014;130:A9473
Abstract
Background: Evidence suggests inferior outcomes among term neonates with serious congenital heart disease delivered at 37 through 38 weeks (early term) gestation compared to their term (39 through 40 weeks) gestation counterparts. We sought to quantify the impact of an early term gestation delivery upon resource demands following neonatal congenital heart surgery.
Methods and Results: We queried The Pediatric Health Information System administrative database for all neonates delivered at term gestation (37 through 40 weeks) undergoing congenital heart surgery at less than 31 days of age from January 2009 through December 2012 classified by the Risk Adjustment in Congenital Heart Surgery (RACHS-1) system. Patients with absent financial data were excluded from consideration. Financial data were adjusted for 2012 dollars. Of 5139 patients meeting inclusion criteria, median birthweight was 3.2 kg (interquartile range [IQR] 2.9-3.5), 60% were male, and 41% were delivered at an early term gestation. Perioperative morbidities more common among early term gestation infants included gastroesophageal reflux (21% v. 17%, p=0.002), acute kidney injury (13% v 11%, p=0.03), and pacemaker placement (1.7% v. 0.9%, p=0.02). Postoperative length of stay was longer among early term gestation infants (18 v. 14 days, p<0.001), with a trend toward higher operative mortality among early term gestation infants in a univariate analysis (7.8% v. 6.4%, p=0.06). Multivariate regression demonstrated that independent of perioperative covariates including birthweight, low center volume, RACHS 6 classification, diagnosis of a genetic syndrome, and government payor class, each early term gestation delivery was associated with a $35748 increase in hospital charges (95%CI $2692-$68803, p=0.034) and a $14118 increase in total estimated hospital cost (95%CI $754-$27482, p=0.038).
Conclusions: We demonstrate and quantify significant and independent increases in resource use among early term gestation infants undergoing congenital heart surgery. Together with existing literature, our findings support the avoidance of elective early term delivery in the setting of prenatally diagnosed congenital heart disease requiring operative interventions in the neonatal period.
Article Information
vol. 130 no. Suppl 2 A9473
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Pediatrics, Vanderbilt Univ Sch of Medicine, Nashville, TN
- 2Pediatric Cardiac Surgery, Vanderbilt Univ Sch of Medicine, Nashville, TN
Abstract 20624: Coronary Revascularization Procedures in Children: A Review of the Pediatric Cardiac Care Consortium
Kalpana Thammineni, Jeffrey Vinocur, Brian Harvey, James St Louis, Lazaros Kochilas
Circulation. 2014;130:A20624
Abstract
Introduction: Coronary revascularization via coronary arterial bypass grafting (CABG) or percutaneous coronary intervention/stents (PCI) is rarely required in children. We report frequency, indications, in-hospital mortality and follow–up of coronary revascularization procedures from a registry of pediatric cardiac centers.
Methods: Retrospective cohort study (1982-2007) from the Pediatric Cardiac Care Consortium for CABG or PCI in children (<18 years). We collected age at procedure, primary diagnosis, indication, timing relative to other procedures, in-hospital mortality, and long term follow up data.
Results: We identified 109 patients with coronary revascularization procedures (males 64, females 45). Of them, 92 were primary CABG (median age 8.4 years, range 3 days – 17.4 years) and 17 primary PCI (median age 10.2 years, range 77 days-17.0 years). In total, 79 were performed for surgical heart diseases and 30 for various medical conditions (e.g. Kawasaki disease, heart transplantation). The most common indications were congenital coronary artery abnormalities (CCAA) (n=24), stenosis as part of supra-aortic stenosis (n=8), and sequelae from arterial switch operation (ASO) (n=22) or other operations involving risk for coronary artery injury (n=19). Late coronary artery compromise was related to prior reimplantation as part of ASO (n=14), procedure for anomalous coronary artery origin (n=9) or after aortic root surgery (n=7). There were 13 deaths, with 12 of them related to rescue revascularization during or shortly after another cardiac procedure. Follow up data existed for 44 of the 97 surviving patients (median time of f/u 24 months, range 1 – 117 months) with documented patency in 22 of them; the remaining patients had partial or total occlusion of the graft or stent with 4 of them needing additional revascularization procedure, 5 receiving heart transplantation, 3 dying at a later time, and 1 needing automatic implantable defibrillator (AICD).
Conclusions: Coronary revascularization procedures can be performed successfully in children after a variety of surgical procedures, CCAA, and medical conditions affecting the coronary arteries. Outcomes are dependent on indication, with the highest mortality in rescue procedures.
Article Information
vol. 130 no. Suppl 2 A20624
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Pediatrics, Univ of Minnesota, Minneapolis, MN
- 2Pediatrics, Univ of Rochester-Golisano Children’s Hosp at Strong, Rochester, NY
- 3Pediatric Surgery, Univ of Minnesota, Minneapolis, MN
- 4Pediatric Cardiac Surgery, Univ of Minnesota, Minneapolis, MN
Abstract 18701: Frequency and Contributing Factors to Diaphragm Paresis Following Pediatric Cardiac Surgery
Intikhab Zafurallah, Osami Honjo, Peter Laussen, Cathy MacDonald, Alejandro Floh
Circulation. 2014;130:A18701
Abstract
Introduction: Diaphragm paresis can occur as a complication of pediatric cardiac surgery that can prolong ventilation and length of ICU stay. Diaphragm plication (DP) may be necessary to improve respiratory mechanics and decrease duration of ventilation support. Early identification of patients who are likely to benefit from DP has not been studied.
Methods: Patients at our institution diagnosed with diaphragm paresis between 2002 – 2012 were identified. Mode of diagnosis, demographics, operative procedures during index admission, and intervals of care were evaluated. Associations between predictors and DP were assessed by univariable and multivariable logistic regressions.
Results: Diaphragm paresis was diagnosed in 161 patients following 6448 index surgeries, of whom 31 (19%) underwent DP (DP+). Paresis was diagnosed by ultrasound in 160 (99%) subjects at a median (IQR) time from surgery of 7 (3, 11) days in DP+ vs 10 (6, 19) days in DP- (p=0.02). DP was completed after a median (IQR) of 4 (1, 17) days after diagnosis. DP+ were younger in age [median (IQR) days DP+ 42 (14, 84) vs DP- 168 (28, 784); p<0.001], underwent surgery of higher RACHS-1 score [DP+ 3 (3, 4) vs DP- 3 (2, 4); p=0.02], and had a higher rate of hypothermic circulatory arrest [DP+ 14 (45%) vs DP- 23 (18%); p=0.001]. DP+ subjects had a rate of single ventricle physiology (32%), median sternotomy (94%), and bypass (87%) similar to DP- subjects. Only younger age (OR 1.003 per day, p=0.02) and use of hypothermic circulatory arrest (OR 3.06, p=0.01) remained significant on multivariable modeling. DP+ subjects had longer duration of ventilator support [DP+ 15 (9, 30) vs DP- 6.5 (3, 12.5) days; p<0.001] and ICU admission [DP+ 23 (18, 42) vs DP- 8 (5, 17) days; p<0.001]. However, ventilation was discontinued after a median of 1 (1,2) day after plication. The time interval from index surgery to diagnosis (EST 0.91, p<0.0001) and interval from diagnosis to DP (EST 0.94, p<0.0001; r2=0.91) were associated with a longer ICU stay even after adjusting for age and bypass time.
Conclusion: Diaphragm paresis is common after congenital heart surgery. Earlier diagnosis and plication may shorten length of ventilation support and ICU stay, particularly in younger patients. Long-term outcome studies following DP are required.
Article Information
vol. 130 no. Suppl 2 A18701
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Intensive Care Medicine, Birmingham Children’s Hosp, Birmingham, United Kingdom
- 2Cardiovascular Surgery, The Hosp for Sick Children, Toronto, Canada
- 3Critical Care Medicine, The Hosp for Sick Children, Toronto, Canada
- 4Diagnostic imaging, The Hosp for Sick Children, Toronto, Canada
Abstract 17696: Pulmonary Vein Obstruction: a European Congenital Heart Surgeons Association (ECHSA) Multicentric Study
David Kalfa, Emre Belli, Emile Bacha, Virginie Lambert, Duccio di Carlo, Heikki Sairanen, Jean Rubay, Martin Kostolny, Christian Schreiber, Matej Nosal, Mark Hazekamp, Hakan Berggren, Illya Yemets, Bohdan Maruszewski, George Sarris, Marco Pozzi, Tjark Ebels, François Lacour-Gayet
Circulation. 2014;130:A17696
Abstract
Pulmonary vein obstruction (PVO) still has a poor prognosis and its optimal management and risk factors for pejorative outcomes remain controversial in the absence of large multicentric studies. We assessed the hypothesis that diffuse PVO and postoperative pulmonary hypertension are associated with worse outcomes while the sutureless surgical technique is associated with better outcomes.
Methods: One hundred and seven patients treated for native or acquired PVO in 16 European or North-American centers (2000-2012) were included retrospectively. A specific PVO severity score (range:0-16) was developed. Endpoints were 1) PVO-related death, 2) persistence or recurrence of PVO, and 3) PV reintervention or PVO-related death. A univariate and multivariate risk analysis (logistic regression and Cox model) was performed. Mean follow-up was 34±41 months.
Results: Patient characteristics, outcomes and risk analyses are summarized in Tables 1, 2 and 3 respectively. In conclusion, both native and acquired PVO still have a poor prognosis. Sutureless repair is significantly associated with lower risks of PVO recurrence and PV reoperation/PVO-related mortality in the univariate analysis. A postoperative pulmonary hypertension and a high degree of severity of the disease evaluated by a new severity score are independent risk factors for pejorative outcomes.
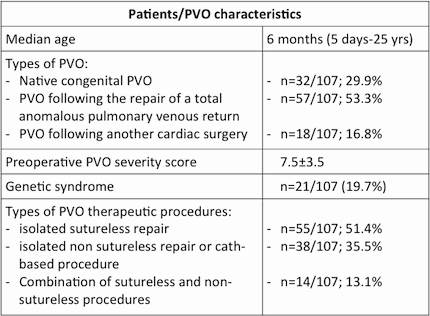
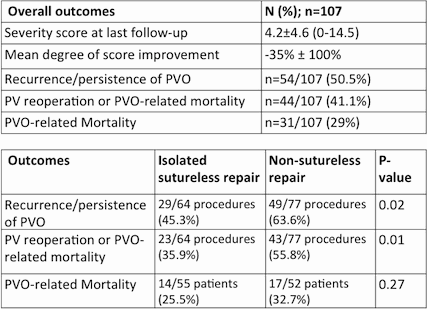
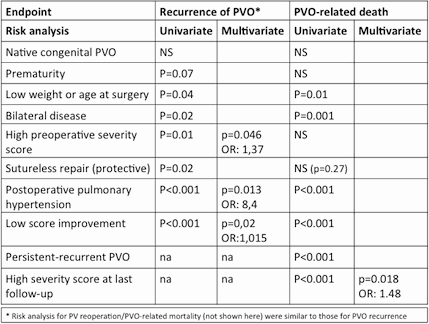
Article Information
vol. 130 no. Suppl 2 A17696
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- David Kalfa1;
- Emre Belli2;
- Emile Bacha3;
- Virginie Lambert4;
- Duccio di Carlo5;
- Heikki Sairanen6;
- Jean Rubay7;
- Martin Kostolny8;
- Christian Schreiber9;
- Matej Nosal10;
- Mark Hazekamp11;
- Hakan Berggren12;
- Illya Yemets13;
- Bohdan Maruszewski14;
- George Sarris15;
- Marco Pozzi16;
- Tjark Ebels17;
- François Lacour-Gayet18,
- European Congenital Heart Surgeons Association (ECHSA)
- 1Pediatric Cardiac Surgery, Marie Lannelongue Hosp, New York-Presbyterian Morgan Stanley Children’s Hosp, New York, NY
- 2Pediatric Cardiac Surgery, Marie Lannelongue Hosp, Paris, France
- 3Pediatric Cardiac Surgery, New York-Presbyterian Morgan Stanley Children’s Hosp, New York, NY
- 4Pediatric Cardiology, Marie Lannelongue Hosp, Paris, France
- 5Pediatric Cardiac Surgery, Ospedale Pediatrico Bambino Gesù, Roma, Italy
- 6Pediatric Cardiac Surgery, Hosp for Children and Adolescents, Univ of Helsinki, Helsinki, Finland
- 7Pediatric Cardiac Surgery, Université Catholique de Louvain, Clinique Universitaires Saint-Luc, Bruxelles, Belgium
- 8Pediatric Cardiac Surgery, Great Ormond Street hospital, London, United Kingdom
- 9Pediatric Cardiac Surgery, German Heart Cntr Munich Clinic of Cardiovascular Surgery at the Technical Univ Munich, Munich, Germany
- 10Pediatric Cardiac Surgery, National Institute of Cardio-Vascular Diseases – Childrens Heart Cntr, Bratislava, Slovakia
- 11Pediatric Cardiac Surgery, Leiden Univ Med Cntr, Leiden, Netherlands
- 12Pediatric Cardiac Surgery, Children’s Heart Cntr, The Queen Silvia Children’s Hosp, Goteborg, Sweden
- 13Pediatric Cardiac Surgery, Ukrainian Childrens Cardiac Cntr, Kyiv, Ukraine
- 14Pediatric Cardiac Surgery, The Children’s Memorial Health Institute, Warsaw, Poland
- 15Pediatric Cardiac Surgery, Athens Heart Surgery Institute, Mitera Pediatric and Hygeia Hosps, Athens, Greece
- 16Pediatric Cardiac Surgery, Ospedali Riuniti, Ancona, Italy
- 17Pediatric Cardiac Surgery, Univ Med Cntr Groningen, Groningen, Netherlands
- 18Pediatric Cardiac Surgery, Royal Brompton Hosp, London, United Kingdom
Abstract 16590: Late Myocardial Infarction After Fontan in Pulmonary Atresia With Intact Ventricular Septum
Adam J Small, Yuli Y Kim, Andrew C Glatz, Kevin K Whitehead, Kiona Y Allen, Tacy E Downing, Richard Donner, Stephanie Fuller, Therese M Giglia
Circulation. 2014;130:A16590
Abstract
Background: Right ventricle (RV)-to-coronary fistulae, coronary stenoses, and atresia are common in pulmonary atresia with intact ventricular septum (PA-IVS). Myocardial infarction (MI) and sudden death after RV decompression in the neonate have been associated with coronary anomalies that create a RV-dependent coronary circulation. There is a paucity of data, however, on long-term coronary sequelae after Fontan completion.
Methods: A retrospective review of patients followed at our institutions who underwent Fontan for PA-IVS between 1985 and 2013 was performed. Patients with antegrade pulmonary blood flow were excluded. Charts were reviewed for sudden death as well as for clinical, radiologic, and angiographic evidence of myocardial infarction/ischemia.
Results: Forty-six patients met inclusion criteria with a median age at Fontan of 2.6 (range 1.2 – 18.9) years. Median follow-up from Fontan was 3.5 (range 0 – 27.9) years. Twenty-three patients (50%) had adequate angiograms for review and of those, 18 (75%) had RV-to-coronary fistulae; 13 (54%) had fistulae with coronary stenoses; and 2 (8.3%) had aortocoronary atresia. There were 2 early deaths (4.3%) but neither had angiograms nor non-invasive tests of myocardial perfusion for review. Five patients (11%) had evidence of ischemia. Of those, 2 (4.3%) had non-fatal MI at 16.9 and 18.8 years post-Fontan. Both patients had fistulae and one had severe mid-right coronary stenosis. Coronary anomalies correlated with ischemic territory on non-invasive testing in both. Three additional patients had evidence of ischemia on stress test and/or cardiac MRI without clinical evidence of MI. Two of those had angiograms, both with fistulae and coronary stenoses that correlated with ischemic territory on non-invasive testing. No patient had traditional risk factors for atherosclerotic coronary disease. No patient underwent coronary revascularization. There was no sustained ventricular tachycardia or sudden death. There were no late deaths.
Conclusion: Patients with PA-IVS and single-ventricle palliation remain at risk for long-term myocardial ischemia. Further study is necessary to define risk factors for ischemic outcomes.
Article Information
vol. 130 no. Suppl 2 A16590
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Adam J Small1;
- Yuli Y Kim1;
- Andrew C Glatz2;
- Kevin K Whitehead2;
- Kiona Y Allen2;
- Tacy E Downing2;
- Richard Donner2;
- Stephanie Fuller3;
- Therese M Giglia2
- 1Internal Medicine, Hosp of the Univ of Pennsylvania, Philadelphia, PA
- 2Pediatrics, The Children’s Hosp of Philadelphia, Philadelphia, PA
- 3Surgery, The Children’s Hosp of Philadelphia, Philadelphia, PA
Abstract 16311: Survival of Fontan Patients After Heart Transplant; Has Survival Improved in the Current Era?
Kathleen E Simpson, James K Kirklin, David C Naftel, Elizabeth Pruitt, Rakesh Singh, R E Edens, Aliessa P Barnes, Charles E Canter
Circulation. 2014;130:A16311
Abstract
Heart transplant (HT) after the Fontan procedure has been associated with increased mortality compared to other pediatric heart diseases. Recent data from the Pediatric Heart Transplant Study demonstrated improved survival after HT over time in children with congenital heart disease (CHD). We hypothesized that this trend extends to Fontan patients undergoing HT in the current era.
Between 1993 to December 2012, 680 pediatric patients who were ≥ 2 years old at listing with CHD underwent HT, of which 254 had a prior Fontan Procedure. Era 1 (1993-2006: 175 Fontan, 376 non-Fontan CHD (NF-CHD)) was compared to Era 2 (2007-2012: 197 Fontan, 221 NF-CHD) for listed patients ≥ 2 years old. An analysis of survival and risk factors by multivariable analysis examined our study hypothesis.
Similar to other groups, Fontan patients in the late era were more likely to be listed as status 1 (69 vs. 90%, p<0.0001), require inotropic support (55 vs. 69%, p=0.0218), have failure to thrive (20 vs. 36%, p=0.0038) and were younger at Fontan (4.3 vs. 3.4 years, p=0.0196) compared to the early era. NF-CHD patients had better survival after HT (p=0.0378) in the late versus early era. Fontan patients failed to show similar improvement in survival between eras (p=0.319). There was a non-significant trend toward improved survival in NF-CHD compared to Fontan patients in the late era (p=0.0607) (Figure). Risk factors (HR, 95% CI) for increased mortality after HT in the Fontan patients by multivariate analysis included longer ischemic time (1.29, 1.03-1.63, p=0.0296) and ventilator dependence (5.41, 2.5-11.72, p<0.0001).
In contrast to the improvements seen in survival among children ≥ 2 years old with other CHD, Fontan patient survival post-HT has not improved. Fontan patients continue to have poorer survival after HT than other CHD. This may be partly explained by the increased risk profile of Fontans at HT in the current era; death being particularly likely when patients are ventilator dependent at transplant.
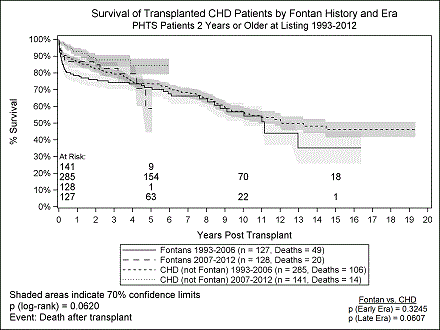
Article Information
vol. 130 no. Suppl 2 A16311
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Kathleen E Simpson1;
- James K Kirklin2;
- David C Naftel2;
- Elizabeth Pruitt2;
- Rakesh Singh3;
- R E Edens4;
- Aliessa P Barnes5;
- Charles E Canter1
- 1Pediatrics, Washington Univ in Saint Louis, Saint Louis, MO
- 2Surgery, Univ of Alabama Birmingham, Birmingham, AL
- 3Pediatrics, Univ of California San Diego, San Diego, CA
- 4Pediatrics, Univ of Iowa, Iowa City, IA
- 5Pediatrics, Univ of Missouri Kansas City, Kansas City, MO
Abstract 15947: In-vitro and Computational Study of the Assisted Bidirectional Glenn Procedure for Initial Palliation of Single Ventricle Physiology
Mahdi Esmaily-Moghadam, Jian Zhou, Tain-Yen Hsia, Richard Figliola, Alison Marsden
Circulation. 2014;130:A15947
Abstract
Introduction: For single ventricle hearts, palliation with systemic-to-pulmonary shunt remains unsatisfactory. However, initial bidirectional Glenn anastomosis is discouraged because the neonatal SVC may be unable to provide adequate pulmonary blood flow (PBF). We propose a radical approach, called the Assisted Bidirectional Glenn (ABG), where flow in a Glenn would be augmented by a shunt between the innominate artery and SVC. Motivated by the ejector pump concept in fluids engineering, we attempt to improve PBF without increasing SVC pressure. ABG is proposed as a replacement for the stage-one operation and potentially can be converted to stage-two circulation by blocking the shunt via a catheter.
Methods: A multiscale framework is adopted to model, both numerically and experimentally, the conventional stage-one (BT shunt) and neonatal stage-two surgeries (Glenn), as well as the ABG. Multiscale modeling allows for prediction of global response of the circulatory system to the altered anatomy, as well as detailed hemodynamics. Lumped parameter network is constructed and tuned based on clinical measurement of 23 stage-one patients. A mock circulatory system was built to experimentally simulate the three surgical options and to validate computational results. Two levels of pulmonary vascular resistance (PVR) are considered to simulate neonatal (high PVR) and pre-stage two (normal PVR) conditions.
Results and Conclusions: Our simulation results show that ABG provides (1) 30% higher PBF compared with stage-two; (2) 14% higher systemic oxygen delivery compared to stage-one; (3) 46% lower heart load compared to stage-one and; (4) 5 mmHg higher SVC pressure compared to stage-two (Figure 1-a). Experimental results verify the simulations, confirming the changes in PBF and SVC pressure (Figure 1-b). Future studies will focus on methods to decrease the SVC pressure with enhanced PBF through design optimization.
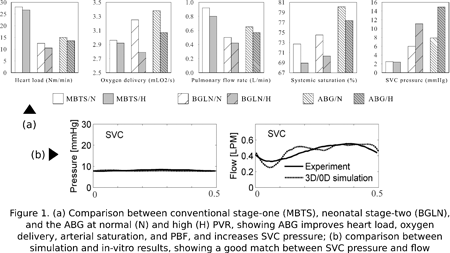
Article Information
vol. 130 no. Suppl 2 A15947
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Mechanical and Aerospace Engineering, Univ of California San Diego, La Jolla, CA
- 2Mechanical Engineering, Clemson university, Clemson, SC
- 3Cadiac Unit, Great Ormond Street Hosp, London, United Kingdom
Abstract 14058: Intramural Ventricular Septal Defects After Repair of Conotruncal Anomalies Are Associated With Postoperative Morbidity
Jyoti K Patel, Reena M Ghosh, Andrew C Glatz, Chitra Ravishankar, Christopher E Mascio, Thomas L Spray, Meryl S Cohen
Circulation. 2014;130:A14058
Abstract
Introduction: Intramural ventricular septal defects (VSDs) are tunnel-like communications between a great artery and right ventricular free wall trabeculations that can occur after repair of conotruncal anomalies. We sought to assess the prevalence of residual intramural VSDs and their impact on postoperative course.
Methods: We reviewed all patients at our institution who underwent biventricular repair of a conotruncal anomaly involving VSD baffle to a great artery from the left ventricle from 1/1/11 to 6/30/13. Perioperative echocardiographic images were reviewed for residual intramural or non-intramural VSDs. The primary outcome was a composite of ECMO use, cardiac transplantation, or mortality. Secondary outcomes included hospital length of stay (LOS) and need for repeat catheter-based or surgical VSD closure.
Results: Of 158 total subjects, 92 (58%) were male and diagnoses were tetralogy of Fallot (n=111), d-transposition of the great arteries (14), truncus arteriosus (13) and other (20). Median age at surgery was 51 days (IQR 6,107). Residual VSD was present in 111 subjects; 29 (26%) had an intramural defect, and 82 (74%) had non-intramural defects (71 peripatch defects, 6 small muscular defects, and 5 with both). Of the non-intramural VSDs, 79 (96%) were hemodynamically insignificant lesions (<2mm). Residual intramural VSDs were more likely than non-intramural VSDs to be greater than 2mm (48% vs 4%, p<0.001). There was no difference in weight, age at operation, or type of conotruncal anomaly based on type of residual VSD. The primary composite outcome was more prevalent in subjects with residual intramural defects compared to non-intramural defects (5/29 [17%] vs 4/82 [5%], p = 0.05). In addition, subjects with residual intramural VSDs had longer LOS (24 days [IQR 13,67] vs 11 days [IQR 5,23], p=0.002). The rates of repeat catheter-based or surgical closure were low overall but higher in subjects with intramural VSDs (8/29 [28%] vs 5/82 [6%], p=0.005).
Conclusions: Intramural VSDs are common after repair of conotruncal anomalies and tend to be larger than non-intramural VSDs. Although most subjects still have favorable outcome, intramural VSDs are associated with greater postoperative morbidity, longer LOS, and need for reintervention.
Article Information
vol. 130 no. Suppl 2 A14058
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Jyoti K Patel;
- Reena M Ghosh;
- Andrew C Glatz;
- Chitra Ravishankar;
- Christopher E Mascio;
- Thomas L Spray;
- Meryl S Cohen
- Div of Cardiology/EP, 8NW, Children’s Hosp of Philadelphia, Philadelphia, PA
Abstract 13663: The Unnatural History of the Ventricular Septal Defect: Outcome up to 40 Years after Surgical Closure
Myrthe E Menting, Judith A Cuypers, Petra Opic, Elisabeth M Utens, Willem A Helbing, Maarten Witsenburg, Annemien E van den Bosch, Ron T van Domburg, Folkert J Meijboom, Ad J Bogers, Jolien W Roos-Hesselink
Circulation. 2014;130:A13663
Abstract
Introduction: Prospective data on long term outcome after ventricular septal defect (VSD) closure do not exist.
Objectives: To evaluate survival and clinical outcome in a prospectively followed patient cohort up to 40 years after surgical VSD closure.
Methods: A cohort of 174 consecutive patients who had surgical VSD repair at young age between 1968-1980, is investigated every 10 years. The study protocol comprised echocardiography, ergometry, Holter monitoring, NT-proBNP, and SF-36 questionnaire.
Results: Survival status was obtained in 90% of 174 patients, and 84% of the eligible survivors participated in the in-hospital examination after median follow-up of 36 (range 30-40) years. A simple concomitant cardiac lesion (non-isolated VSD) was present in 32%. Postoperative mortality within 30 days was 10%, and late mortality at 40 years was 14%. Eight patients died in the last decade: 3 cardiac (arrhythmia n=2, heart failure n=1), 3 non-cardiac, and 2 unknown causes. Cumulative event incidence at 40 years was 38%, including interventions in 13%, symptomatic arrhythmias in 11%, and pacemaker/ICD in 7%. Most events occurred in non-isolated VSD patients (Fig. 1). LV systolic function was impaired in 21%, but remained stable over the last decade. Prevalence of RV systolic dysfunction increased from 1% to 16% (p=0.001). Mean workload decreased from 91% to 87% of expected (p=0.01). NT-proBNP (median 11.6 [IQR 7.0-19.8] pmol/L) was elevated in 38%. Postoperative complete atrioventricular block and non-isolated VSD were predictive for late events (HR 3.9 [95%CI 1.2-13.0]; HR 3.5 [95%CI 1.7-7.2]). Regarding the SF-36, patients scored their health status significantly better than the reference population.
Conclusions: Survival up to 40 years after successful surgical VSD closure is slightly lower than in the general Dutch population. Although many patients are discharged from routine follow-up, morbidity is substantial, especially in patients with non-isolated VSD.
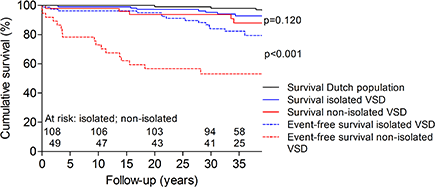
Article Information
vol. 130 no. Suppl 2 A13663
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Myrthe E Menting1;
- Judith A Cuypers1;
- Petra Opic1;
- Elisabeth M Utens2;
- Willem A Helbing3;
- Maarten Witsenburg1;
- Annemien E van den Bosch1;
- Ron T van Domburg1;
- Folkert J Meijboom4;
- Ad J Bogers5;
- Jolien W Roos-Hesselink1
- 1Cardiology, Erasmus MC, Rotterdam, Netherlands
- 2Psychiatry and Psychology, Erasmus MC, Rotterdam, Netherlands
- 3Cardiology, Erasmus MC – Sophia Children’s Hosp, Rotterdam, Netherlands
- 4Cardiology, Univ Med Cntr Utrecht, Utrecht, Netherlands
- 5Cardiothoracic surgery, Erasmus MC, Rotterdam, Netherlands
Abstract 11837: A Vascular Endothelial Growth Factor a (VEGFA) Genetic Variant is Associated With Improved Ventricular Function and Transplant-Free Survival After Surgery for Non-Syndromic Congenital Heart Defects
Constantine D Mavroudis, Daniel S Kim, Nancy Burnham, Alexandra H Morss, Jerry H Kim, Amber Burt, David R Crosslin, Donna M McDonald-McGinn, Elaine H Zackal, Meryl S Cohen, Susan C Nicolson, Thomas L Spray, Ian B Stanaway, Deborah A Nickerson, Mark W Russell, Hakon Hakonarson, Gail P Jarvik, J William Gaynor
Circulation. 2014;130:A11837
Abstract
Introduction: We have shown the minor allele of VEGFA single nucleotide polymorphism (SNP) rs833069 is associated with improved transplant-free survival after surgery for CHD in infants. The biological mechanisms underlying the improved survival have not been delineated. The minor allele is associated with higher VEGF production. We hypothesized that the VEGFA SNP is associated with better systemic ventricular function after surgery for CHD, resulting in improved survival.
Methods: This is an analysis of a cohort of 422 non-syndromic CHD patients who underwent cardiac surgery with cardiopulmonary bypass before 6 months of age, of which 105 (24.9%) were single ventricle. Echocardiography reports were reviewed. Systemic ventricular function was subjectively assessed and categorized as normal, or mildly, moderately or severely depressed. The change in ventricular function was calculated as the change from the preoperative study to last available study. Stepwise linear regression, adjusting for covariates, was performed for the outcome of change in ventricular function. Model comparison was performed using Akaike’s information criterion (AIC). Only variables that improved model prediction of change in systemic ventricular function were retained in the final model.
Results: Genetic and echocardiographic data were available for 335/422 subjects (79%). Of these, 40 (11.9%) developed worse systemic ventricular function and none demonstrated an improvement in ventricular function during the study period. (mean followup 13.5 years). After covariate adjustment, presence of the VEGFA SNP rs833069 minor allele was associated with preserved ventricular function, (p=0.011). A decrease in systemic ventricular function was significantly associated with heart transplantation or death (p<0.01).
Conclusions: These data are consistent with the hypothesis that the mechanism by which the VEGFA SNP rs833069 minor allele improves survival may be preservation of ventricular function. Further studies are needed to validate this genotype-phenotype association and determine if the mechanism underlying preservation of ventricular function and improved transplant-free survival is related to increased VEGF production.
Article Information
vol. 130 no. Suppl 2 A11837
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Constantine D Mavroudis1;
- Daniel S Kim2;
- Nancy Burnham1;
- Alexandra H Morss1;
- Jerry H Kim3;
- Amber Burt2;
- David R Crosslin2;
- Donna M McDonald-McGinn4;
- Elaine H Zackal4;
- Meryl S Cohen5;
- Susan C Nicolson6;
- Thomas L Spray1;
- Ian B Stanaway7;
- Deborah A Nickerson7;
- Mark W Russell8;
- Hakon Hakonarson9;
- Gail P Jarvik2;
- J William Gaynor1
- 1Cardiothoracic Surgery, Children’s Hosp of Philadelphia, Philadelphia, PA
- 2Med Genetics, Univ of Washington Sch of Medicine, Seattle, WA
- 3Anesthesia, Seattle Children’s Hosp, Seattle, WA
- 4Genetics, Children’s Hosp of Philadelphia, Philadelphia, PA
- 5Cardiology, Children’s Hosp of Philadelphia, Philadelphia, PA
- 6Cardiothoracic Anesthesiology, Children’s Hosp of Philadelphia, Philadelphia, PA
- 7Genome Sciences, Univ of Washington Sch of Medicine, Seattle, WA
- 8Pediatric Cardiology, C.S. Mott Children’s Hosp, Univ of Michigan, Ann Arbor, MI
- 9Applied Genomics, Children’s Hosp of Philadelphia, Philadelphia, PA
Abstract 11506: Total Cavopulmonary Connection Geometry and Hemodynamics: Implications on Patient Exercise Performance
Elaine Tang, Reza H Khiabani, Kevin K Whitehead, Maria Restrepo, Lucia Mirabella, James Bethel, Stephen M Paridon, Bradley S Marino, Mark A Fogel, Ajit P Yoganathan
Circulation. 2014;130:A11506
Abstract
Background: Single ventricle (SV) patients with a total cavopulmonary connection (TCPC) have limited exercise capacity. Elevated TCPC energy loss is hypothesized to decrease SV efficiency and impede exercise performance. Optimizing TCPC surgical design holds the promise of improving exercise capacity. This study aimed to investigate the correlations between TCPC geometry features, TCPC exercise energy loss, and patient exercise performance.
Methods: Forty seven TCPC patients (age=19±6years) who completed standard exercise stress test and exercise cardiac magnetic resonance (CMR) were included. TCPC anatomies were reconstructed from CMR images and geometric features analyzed. Vessel flows at ventilatory anaerobic threshold (VAT) were obtained from phase contrast CMR images acquired immediately following lower leg exercise using a cycle ergometer. Numerical simulations were performed to quantify TCPC power loss during exercise (iPLVAT). Regressions between TCPC geometry features, iPLVAT, and minute oxygen consumption at VAT (VO2) were investigated.
Results: Significant correlations are shown in Figure 1. VO2 was negatively correlated with age. In adolescents (12-18yrs), VO2 was negative correlated with iPLVAT and positively correlated with normalized minimum TCPC diameter. For adults (>18yrs), these correlations were not significant. Instead, VO2 was positively correlated with normalized total systemic venous return, and negatively correlated with age.
Conclusions: In adolescent Fontan patients, lower normalized minimum TCPC diameter and elevated power loss may contribute to worse exercise performance. Alternatively, exercise performance in adult patients worsens with increasing age and reduced systemic venous return. Understanding these relationships will allow clinicians to identify Fontan patients at risk for diminished exercise tolerance and may allow for interventions to maximize patient exercise performance.
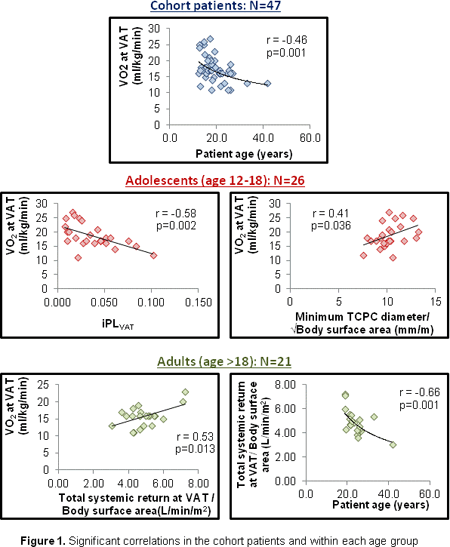
Article Information
vol. 130 no. Suppl 2 A11506
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Elaine Tang1;
- Reza H Khiabani1;
- Kevin K Whitehead2;
- Maria Restrepo1;
- Lucia Mirabella1;
- James Bethel3;
- Stephen M Paridon2;
- Bradley S Marino4;
- Mark A Fogel2;
- Ajit P Yoganathan1
- 1Biomedical Engineering, Georgia Institute of Technology, Atlanta, GA
- 2Div of Cardiology, Children’s Hosp of Philadelphia, Philadelphia, PA
- 3BDMC, Westat, Inc., Rockville, MD
- 4Heart Institute, Cincinnati Children’s Hosp Med Cntr, Cincinnati, OH
Abstract 19383: Impact of Tight Glycemic Control on the Neurodevelopmental Outcomes at One Year of Age of Children with Congenital Heart Disease
Anjali Sadhwani, Lisa Asaro, Caren Goldberg, Janice Ware, Jennifer Butcher, David Wypij, Michael Gaies, Cynthia Smith, Jamin Alexander, Michael Agus
Circulation. 2014;130:A19383
Abstract
Background: While postoperative tight glycemic control (TGC) may reduce morbidity in certain subgroups of children undergoing cardiac surgery, its association with later neurodevelopmental outcomes is not well-characterized. Both hypoglycemia and hyperglycemia impact brain development after pediatric cardiac surgery. The aim of this study was to assess the association of post-operative TGC in children undergoing cardiac surgery with neurodevelopmental outcomes at one year of age.
Methods: A two-center, prospective, randomized trial of TGC vs. standard care (STD) was conducted in 980 children with congenital heart disease, from 0 to 36 months of age, undergoing cardiopulmonary bypass surgery. Subjects were eligible for one-year follow-up evaluations if they were less than one year of age when enrolled in the trial and were born after March 1, 2008 (N=681). In-person testing was conducted using the Bayley Scales of Infant Development, Third Edition (BSID-III). Parents completed the Adaptive Behavior Assessment System, Second Edition (ABAS-II) to assess adaptive functioning.
Results: Of 681 eligible patients, 200 patients (TGC=100, STD=100) returned for neurodevelopmental testing and 166 parents completed the ABAS-II. Mean age at testing was 13 months (TGC 13.2±1.8, STD 13.3±1.8). Nineteen percent of children assigned to TGC and 15% assigned to STD had a known genetic anomaly. For the remaining 166 patients without a known genetic anomaly, cognitive composite scores (103.9±12.8 in the combined groups) on the BSID-III fell in the Average range while language (97.0±12.1) and motor (92.2±14.2) composite scores and overall adaptive functioning on the ABAS-II [(Global Adaptive Composite (GAC) 91.5±14.0] were significantly lower than population norms (p<0.001). On the ABAS-II, 28% of the patients had concerns (scores< 1 SD below the mean) in the health & safety domain, 35% in the motor domain and 47% in the self-care domain. No significant treatment group differences were found in the BSID-III [cognitive (p=0.60), language (p=0.66), or motor (p=0.29)] or ABAS-II [GAC (p=0.15)] composite scores.
Conclusion: For children undergoing congenital heart surgery, post-operative TGC is not associated with neurodevelopmental outcomes at one year of age.
Article Information
vol. 130 no. Suppl 2 A19383
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Anjali Sadhwani1;
- Lisa Asaro2;
- Caren Goldberg3;
- Janice Ware4;
- Jennifer Butcher3;
- David Wypij2;
- Michael Gaies3;
- Cynthia Smith5;
- Jamin Alexander6;
- Michael Agus7
- 1Psychiatry, Boston Children’s Hosp, Boston, MA
- 2Cardiology, Boston Children’s Hosp, Boston, MA
- 3Pediatrics, C.S. Mott Children’s Hosp, Ann Arbor, MI
- 4Developmental Medicine, Boston Children’s Hosp, Boston, MA
- 5Cardiology, C.S. Mott Children’s Hosp, Ann Arbor, MI
- 6Critical Care Medicine, Boston Children’s Hosp, Boston, MA
- 7Medicine Critical Care, Boston Children’s Hosp, Boston, MA
Abstract 18729: Comparative Analysis of the Use of Extracorporeal Membrane Oxygenation During Cardiopulmonary Resuscitation Within a Pediatric Cardiac Intensive Care Unit
Stephanie J Conrad, Matthew K Bacon, Brittney C Hatch, John David Hughes, Michelle K Terrell, Patrick Maynord, Andrew H Smith
Circulation. 2014;130:A18729
Abstract
Background: Mechanical support to aid in restoration of circulation during cardiopulmonary resuscitation (CPR) is increasingly common in the pediatric cardiac intensive care unit (CICU). We sought to both identify and quantify factors predicting the implementation of extracorporeal membrane oxygenation to support CPR (eCPR).
Methods and Results: Events associated with CPR from July 2010 through December 2013 within our pediatric CICU were retrospectively reviewed. Of 135 arrests among 88 patients, 84% were among postsurgical patients and 98% (n=133) resulted in a return of circulation, either spontaneous (n=100, 74%) or with the assistance of mechanical support (n=33, 24%). Median age at arrest was 106 days (interquartile range [IQR] 26-207 days) and weight was 3.9 kg (IQR 3.0-6.0 kg). Median length of stay (LOS) at the time of arrest was 5 days (IQR 1-49 days). Common primary causes included low cardiac output (38%), respiratory failure (33%), and arrhythmia (15%). Univariate predictors of an eCPR arrest included smaller size (3.3 v. 4.3 kg, p=0.004), younger age (25 v. 130 days, p<0.001), shorter length of stay at time of arrest (1 v. 8.5 days, p=0.001), single ventricle physiology (30% v. 14% among biventricular physiology arrests, p=0.04), and arrests not related to respiratory failure (34% v. 5% eCPR among respiratory failure arrests, p<0.001). Unit factors not associated with an increased frequency of arrests resulting in eCPR included unit capacity, night shift, and the experience levels of both the bedside nurse and attending. Among patients with at least one arrest, median ICU LOS was 18 days (IQR 9-72 days) and overall survival to ICU discharge was 72%. Survival to ICU discharge was not significantly different with respect to use of eCPR as compared to conventional CPR (60% v. 77% respectively, p=0.11).
Conclusions: We report predictors of the need for mechanical support during cardiopulmonary resuscitation within a pediatric CICU, and demonstrate comparable post-resuscitation survival to ICU discharge among those rescued with eCPR. Further longitudinal investigation is necessary to identify potential eCPR-associated differences in morbidity and neurocognitive outcomes following a CICU arrest.
Article Information
vol. 130 no. Suppl 2 A18729
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Stephanie J Conrad;
- Matthew K Bacon;
- Brittney C Hatch;
- John David Hughes;
- Michelle K Terrell;
- Patrick Maynord;
- Andrew H Smith
- Pediatrics, Vanderbilt Univ Sch of Medicine, Nashville, TN
Abstract 17965: Echocardiographic Assessment of Surgically Placed Melody Valves in the Mitral Position in Young Children
Lindsay R Freud, Sitaram M Emani, Audrey C Marshall, Gerald R Marx, Wayne Tworetzky
Circulation. 2014;130:A17965
Abstract
Introduction: Mitral valve replacement (MVR) in young children is limited by lack of small prostheses, resulting in high re-intervention rates and mortality. Therefore, in 2010, we began performing MVR with modified, surgically placed, stented jugular vein grafts (Melody valve) to allow for tailoring to small annular dimensions as well as subsequent expansion in the catheterization lab. We aimed to analyze key pre- and post-operative (op) echo variables to refine surgical planning for this novel procedure.
Methods: We included 17 patients who underwent surgically placed Melody MVR. The pre- and post-op echoes before discharge were retrospectively reviewed for anatomic and physiologic variables. On the pre-op echo, we performed “potential” mitral annular measurements in orthogonal planes to estimate the maximum intra-op balloon sizing. Given concern for left ventricular outflow tract obstruction (LVOTO), a ratio of the narrowest subaortic region in systole to the actual mitral annular dimension (SubA:MV) was assessed in the parasternal long axis view. Values are presented as median (range), where applicable.
Results: Melody MVR was performed at 8 months of age (weight 5.2 kg) for stenosis (5), regurgitation (1), and mixed disease (11). On pre-op echo, the potential mitral measurement in the lateral dimension correlated well with the intra-op balloon size selected by the surgeon (ρ=0.58, p=0.01). The Melody valve was expanded to 1.0 times the potential lateral dimension (median 14 mm). The SubA:MV measured >0.5 in all patients. By post-op day 10 (5-35), the mean mitral gradient improved by 9 mm Hg (1-16), and right ventricular pressure was <½ systemic in 76% (vs. 12% pre-op, p<0.001). Only 2 patients had mild regurgitation and 1 had a mild perivalvar leak. The internal diameter of the Melody valve measured 3.4 mm (1-6) less than the intra-op balloon size. No patient had LVOTO >25 mm Hg or pulmonary vein obstruction.
Conclusion: Intra-op balloon sizing of Melody MVR may be guided by pre-op echo indices to achieve adequate mitral valve function without significant LVOTO. The post-op Melody valve diameter was found to measure less than the intra-op balloon size, which should be considered as experience with the Melody in native, non-conduit positions evolves.
Article Information
vol. 130 no. Suppl 2 A17965
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Dept of Cardiology, Boston Children’s Hosp, Boston, MA
- 2Dept of Cardiac Surgery, Boston Children’s Hosp, Boston, MA
Abstract 17406: Association of Post-operative Complications with Clinical Outcomes and Hospital Costs following the Norwood Operation
Kimberly E McHugh, Sara K Pasquali, Matthew A Hall, Mark A Scheurer
Circulation. 2014;130:A17406
Abstract
Introduction: Patients undergoing the Norwood operation consume considerable healthcare resources. However, data are limited regarding factors impacting hospitalization costs for these patients. We evaluated the association of post-operative complications with hospital costs.
Methods: We utilized a unique dataset consisting of prospectively collected clinical outcomes and complications data from the Pediatric Heart Network Single Ventricle Reconstruction trial linked at the patient level with cost data for hospitals (n=10) participating in the Children’s Hospital Association administrative databases during the trial period. Probabilistic matching of indirect identifiers was used to link records. Cost-to-charge ratios were used to estimate costs, which were adjusted for regional differences and inflation. The relationship between complications and cost was modeled using linear regression, accounting for the skewed distribution of cost, and adjusting for within-center clustering and patient characteristics.
Results: A total of 334 eligible Norwood records (95%) were matched between datasets. Overall, 82% suffered at least one complication (median 2; range 0-33). The most common complications associated with increased hospital costs included: cardiopulmonary resuscitation (22%), arrhythmia (22%), sepsis (15%), and extracorporeal membrane oxygenation (11%). Those with complications had longer post-operative length of stay (25d vs. 12d, p<0.0001), more total ventilator days (7d vs. 5d, p<0.0001), and higher in-hospital mortality (17.6% vs. 3.4%, p<0.01). Adjusted hospital costs in those with a complication were $113,505 (95% CI $99,326 – $129,707) vs. $71,453 (95% CI $59,200 – $86,243) in those without a complication, p=0.0001, and costs increased with the number of complications (1-2 complications = $117,137 vs. 3-4 complications = $155,500 [p<0.01] vs. ≥5 complications $266,386 [p< 0.0001]).
Conclusions: This unique dataset of merged clinical trial and cost data demonstrated that postoperative complications are common following the Norwood operation and are associated with worse clinical outcomes and higher costs. Efforts to reduce complications in this population may lead to both improved outcomes and cost savings.
Article Information
vol. 130 no. Suppl 2 A17406
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Pediatrics, Med Univ of South Carolina, Charleston, SC
- 2Pediatrics, Univ of Michigan CS Mott Children’s Hosp, Ann Arbor, MI
- 3Biostatistics, Children’s Hosp Association, Overland Park, KS
Abstract 16858: Factors Associated With Length of Stay and Cost of the Fontan Surgery: A Multi-Institutional Study
Shaji C Menon, Kevin Hinkle, Molly McFadden, Jacob Wilkes, L. LuAnn Minich
Circulation. 2014;130:A16858
Abstract
Introduction: We sought to determine factors associated with length of stay (LOS) and cost of the Fontan surgery in a contemporary large multi-institutional cohort.
Hypothesis: Independent and modifiable risk factors associated with longer LOS and higher cost will be identified.
Methods: We performed a retrospective cohort study of Fontan surgey (1/2004 – 7/2013) using the Pediatric Health Information Systems (PHIS) database (40 US children’s hospitals). Outcome parameters were LOS and costs at Fontan surgery. Predictor variables were demographics (race, insurance, region, rural vs city, commute time, surgical volume), cardiac and other diagnoses, Glenn events, readmission between Glenn and Fontan, and age at Glenn and Fontan. Following a stepwise selection procedure, significant predictors of LOS and cost were included in a generalized linear model with the appropriate inverse Gaussian distribution and the log link function with LOS and cost (adjusted to 2012 $ and region) as the dependent variable.
Results: For the 2187 patients (62% male) who had Fontan surgery, the median age at surgery was 1146 days (IQR: 943-1369) and at Glenn was 165 days (IQR: 133-208). Following Fontan, the median ICU LOS was 3 days (IQR: 2-6) and total LOS was 9 days (IQR: 7-14). The median total hospital cost was $93, 900 (IQR: 67,800-136,100). Post-operative mortality was 1% (N=21). Region, insurance, race, surgical volume were the predominant factors associated with both cost and LOS (Table 1).
Conclusion: This is the largest multicenter contemporary study describing the factors (particularly healthcare delivery factors) associated with resource utilization during the Fontan hospitalization. Health care delivery variables dominated the factors predicting increased resource utilization. Changes in health care policy should target health care delivery risk factors (race, region, insurance, driving distance, surgical volume) to reduce cost in this resource intensive population.
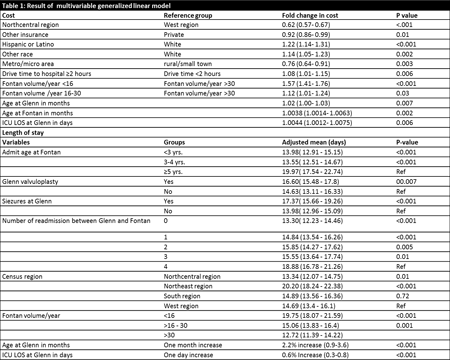
Article Information
vol. 130 no. Suppl 2 A16858
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Shaji C Menon;
- Kevin Hinkle;
- Molly McFadden;
- Jacob Wilkes;
- L. LuAnn Minich
- Pediatrics, Univ of Utah, Salt Lake City, UT
Abstract 16733: High Death and Re-Intervention Rate in Neonatal Modified Blalock-Taussig Shunts With Concomitant Patent Dutus Artertiosus: Mechanistic Insights Into Competitive Flow and Pulmonary Overcirculation
Bari Murtuza, Madhi Esmaily Moghadam, Alison Marsden
Circulation. 2014;130:A16733
Abstract
Background: When palliating neonates with cyanotic heart defects with systemic-pulmonary (SP) shunts, leaving the ductus arteriosus open may lead to competitive flow and/or pulmonary overcirculation. Competitive flow can cause early shunt obstruction. Pulmonary overcirculation can result in poor oxygen delivery. We reviewed the clinical outcomes in neonates following SP shunts in our institution, and conducted multiscale modeling to assess the fluid dynamics of SP shunt with concomitant PDA.
Methods: Retrospective analysis of 69 neonates over a 7-year period was performed: 23 patients had a SP shunt and PDA (MS), and 46 patients had an isolated SP shunt (SS) with PDA ligation. Nearly all patients received modified Blalock-Taussig shunts (MBTS). Patients following Norwood procedure were excluded. Median postoperative PDA diameter was 2.7mm in MS patients. Primary endpoints of death and shunt intervention were assessed. Following the clinical analysis, multiscale models of a 3.5mm MBTS with and without a PDA were constructed. Simulations examine flow dynamics, and predict clinically relevant outcomes such as oxygen delivery.
Results: There was a significantly higher incidence of shunt thrombosis within 3 months in MS vs. SS patients (34.8% vs. 2.2%; p=0.0004). Moreover, the combined endpoint of 30-day mortality/shunt intervention was significantly higher in the MS patients (16% vs. 6%; p<0.01).
Computational simulations showed that in patients with a 3.5mm MBTS and a PDA, despite higher Qp:Qs and systemic oxygen saturation, systemic and coronary oxygen deliveries were lower than shunt only patients. There is also reduced flow velocity magnitude and increased particle residence time within the MBTS (Fig).
Conclusions: When a PDA is left open following SP shunt construction in a neonate, worse clinical outcomes occur. Computer simulations suggest that both pulmonary overcirculation and competitive flow characteristics may contribute to this observation.
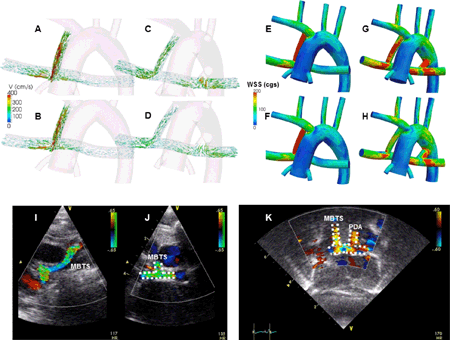
Article Information
vol. 130 no. Suppl 2 A16733
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Cardiothoracic Surgery, Freeman Hosp, Newcastle upon Tyne, United Kingdom
- 2Dept of Mechanical and Aerospace Engineering, UCSD, La Jolla, CA
Abstract 12872: External Stenting for Tracheobronchomalacia Associated With Congenital Heart Disease
Yuki Nakamoto, Makoto Ando, Hisaya Hasegawa
Circulation. 2014;130:A12872
Abstract
Introduction: Tracheobronchomalacia (TBM) is a disease of symptomatic obstruction of tracheal or bronchial portions either related to congenital instability or secondary to vascular compression. Patients with TBM associated with congenital heart disease (CHD) often remain difficult to manage after cardiac surgery, requiring prolonged artificial respiratory support. Under these circumstances, several treatments such as aortopexy, internal stenting, or high positive end-expiratory pressure ventilation, have been attempted, although satisfactory results have not been obtained. In 2008, we started the external stenting technique using a ringed expanded polytetrafluoroethylene graft, and we have treated 39 patients with this technique. We review the results of our experience with external stenting for TBM in children.
Methods: Clinical records of 42 procedures in 39 patients with TBM, who underwent external stenting in our institute from February 2008 to March 2014, were reviewed. The diagnosis of TBM was made by bronchoscopy or enhanced computed tomography.
Results: Etiology of the tracheobronchial obstruction included primary TBM in 10 cases (25.6%), congenital vascular compression in 14 (35.9%), and postoperative vascular compression in 15 (38.5%). Preoperatively, 30 patients (76.9%) were failure to wean from ventilation, 9 (23.1%) were performed tracheotomy, and 5 (12.8%) were performed aortopexy. Forty-six stents were placed in 39 children, 22 boys and 17 girls, with a median age of 7 months (range, 1 months – 6 years). Sixteen stents were placed in the trachea, 25 were placed in the left main bronchus, and 5 were placed in the right main bronchus. Stenosis-free tracheobronchial expansion was achieved in 38 (97.4%) patients. Thirty five (89.7%) patients were successfully weaned from respirator, and 36 (92.3%) discharged from a hospital. Complications of the operation occurred in 4 (9.5%) of the 42 procedures. One patient died of a perioperative bleeding. Three patients were necessitated their stent removals due to bronchial perforation, bronchial stenosis, and pulmonary venous obstruction.
Conclusions: External stenting seems to be extremely useful in the treatment of intractable TBM associated with CHD.
Article Information
vol. 130 no. Suppl 2 A12872
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Pediatric cardiology, Sakakibara Heart Institute, Tokyo, Japan
- 2Cardiovascular surgery, Sakakibara Heart Institute, Tokyo, Japan
- 3Naonatal Intensive Care, Tokyo Women’s Med Univ Med Cntr East, Tokyo, Japan
Abstract 11317: Outcomes of Atrial Arrhythmia Surgery for Atrial Tachyarrhythmias in Patients With Repaired Tetralogy of Fallot
Jiwoon Chang, Sajan Patel, Tristan R Grogan, Jamil A Aboulhosn
Circulation. 2014;130:A11317
Abstract
Background: Atrial tachyarrhythmia is common in adults with tetralogy of Fallot (TOF) due to surgical scarring from repairs and atrial enlargement. The maze procedure refers to surgical ablation within the right atrium to disrupt arrhythmogenic circuits and is sometimes performed concomitantly during reoperation on repaired TOF patients. Our study aims to evaluate the effectiveness of maze in TOF patients.
Methods: We performed a retrospective chart review that identified 30 TOF patients who underwent a pulmonary valve replacement (PVR) with maze and 38 TOF patients who underwent a PVR without maze from 1994 to 2011 and had at least 2 years of post-surgical follow-up at the Ahmanson/UCLA Adult Congenital Heart Disease Center. Preoperative and postoperative arrhythmia status and management were compared in maze and non-maze groups.
Results: Before the procedure, the most common pre-operative arrhythmias in the maze group were a history of atrial fibrillation (AFib)(n=16), atrial flutter (AFL)(n=10), and other supraventricular tachycardia (SVT)(n=6). Isolated right atrial maze was performed in 26 patients, and combined right and left atrial maze-cox procedure was performed in 4 patients. Of the 16 patients in the maze group with pre-op Afib, 6 had recurrent Afib within the first 2 years of follow-up (62.5% relative reduction, p=0.012). Of the 10 patients with pre-op AFL, only 1 had recurrence (90% relative reduction, p=0.012). Of the 6 patients with pre-op SVT, 4 had recurrence (33.3% relative reduction, p=0.727). There was no significant arrhythmia status change in the non-maze group at 2 years. Comparing patients with and without maze, the average cardiopulmonary bypass times were 155 minutes and 97 minutes, respectively (p=0.064), and aortic cross clamp times were 122 minutes and 64 minutes, respectively (p=0.004). On average, patients with maze spent 3.7 more days in the hospital compared to those without maze (p=0.001).
Conclusions: Performing a concomitant maze procedure in patients undergoing TOF repair was associated with a moderate improvement in atrial tachyarrhythmia burden over 2 years. TOF patients who had the concomitant maze procedure required longer cardiopulmonary bypass time, aortic cross clamp time, and total hospital stay.
Article Information
vol. 130 no. Suppl 2 A11317
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Divison of Cardiology, David Geffen Sch of Medicine at UCLA, Los Angeles, CA
- 2Internal Medicine, Ronald Reagan UCLA Med Cntr, Los Angeles, CA
- 3Medicine Statistics Core, David Geffen Sch of Medicine at UCLA, Los Angeles, CA
- 4Cardiology, Ahmanson/UCLA Adult Congenital Heart Disease Cntr, Los Angeles, CA
Abstract 11345: Resource Demands and the Incidence of Cardiopulmonary Resuscitation Within a Pediatric Cardiac Intensive Care Unit
Matthew K Bacon, Stephanie J Conrad, Brittney C Hatch, John D Hughes, Michelle K Terrell, Patrick O Maynord, Andrew H Smith
Circulation. 2014;130:A11345
Abstract
Background: Econometric evidence suggests exponential declines in the ability to provide critical care services as variable demands exceed a relatively fixed supply of available critical care resources. We hypothesized that increasing demands upon resources within a pediatric cardiac intensive care unit (CICU) is also associated with increases in the incidence and rate of cardiopulmonary resuscitation.
Methods and Results: Records from each twelve-hour nursing shift within an eighteen-bed pediatric CICU from 1 July 2010 through 30 April 2014 were retrospectively reviewed. There were 2716 reports available for review from 2769 shifts (97%). During the study period, there were 1,803 surgical and 1,215 medical admissions, accounting for a median census of 15 (interquartile range [IQR] 13-17) patients per shift, and a total of 20,269 patient days (40,538 patient-shifts) over the 44-month study period. Median bed capacity was 83% (IQR 72-94%), and median patient to nursing assignment ratio was 1.5 (IQR 1.4-1.6 patients per nurse assignment). Cardiac arrest (defined as administration of chest compressions) was identified in 138 occasions in 134 shifts, an arrest rate of 3.4 arrests per 1000 patient-shifts. Arrests were no more frequent during night versus day shifts (3.2 v. 3.6 per 1000 patient shifts, p=0.40), nor were they greater during weekend versus weekday shifts (2.9 v. 3.9 per 100 patient-shifts, p=0.14). There was a trend toward an increase in the incidence of cardiac arrest with patient to nurse assignment ratios of less than 1.5 (2.8 v. 3.9 arrests per 1000 patient shifts, p=0.06). Unit occupancy exceeding 85% was associated with a 45% greater rate of cardiac arrest (2.6 v 4.1 arrests per 1000 patient-shifts, 95%CI 0.3 to 2.6 increase, p=0.01).
Conclusions: We report a significant increase in the incidence of cardiopulmonary resuscitation at times of greater resource consumption within a pediatric CICU as defined by unit capacity. Multi-institutional studies are necessary to identify generalizable organizational characteristics that may promote efficient allocation of resources and optimize delivery of care to a population of patients at greater risk for significant hospital morbidity.
Article Information
vol. 130 no. Suppl 2 A11345
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Matthew K Bacon1;
- Stephanie J Conrad1;
- Brittney C Hatch2;
- John D Hughes2;
- Michelle K Terrell2;
- Patrick O Maynord1;
- Andrew H Smith1
- 1Pediatrics, Vanderbilt Univ Sch of Medicine, Nashville, TN
- 2Monroe Carell Jr. Children’s Hosp at Vanderbilt, Vanderbilt Univ Med Cntr, Nashville, TN
Abstract 19663: Failed Delayed Sternal Closure Following Neonatal Cardiac Surgery Predicted by High Mean Airway Pressure and Associated with Increased Post-operative Mortality
Dashiell Massey, Kathryn A Williams, Ravi R Thiagarajan, Frank Pigula, Catherine K Allan
Circulation. 2014;130:A19663
Abstract
Background: Myocardial edema, increased lung water, and anasarca are common following neonatal cardiac surgery with cardiopulmonary bypass and amplify the risk of hemodynamic instability and inadequate ventilation following sternal closure. Delayed sternal closure (DSC) in the intensive care unit one or more days following surgery is a common strategy to mitigate this risk, but has been associated with increased risk of infection. In addition, failed DSC has previously been identified as a risk factor for mortality. This study sought to identify predictor variables and determine impact of failed DSC.
Methods: Records of all neonates undergoing DSC in the cardiac intensive care unit (CICU) following surgery with cardiopulmonary bypass between January 2008 and May 2013 were reviewed. Pre-operative, intra-operative and post-operative variables were compared for those patients who failed DSC versus those who did not. Continuous variables were compared utilizing Wilcoxon’s test and categorical variables using Fisher’s exact test.
Results: Of 256 neonates undergoing DSC in the CICU, 22 failed first attempt at DSC. No significant difference between the two groups was appreciated in age, weight, or bypass (cross clamp, circulatory arrest, and total) times. Comparing DSC failures to successes, significantly more failures: followed Stage I palliation (63% vs. 31%); occurred later (post-operative day 4.7 vs. 2.8, p = 0.009); and were proceeded by higher mean airway pressures (9 vs. 8 cm H2O, p = 0.04), peak inspiratory pressure (27 vs. 24, p = 0.002), and inotrope score (12.1 vs. 9.6, p = 0.06). There was no association with systolic blood pressure or lactate prior to DSC. Failed DSC was associated with increased duration of mechanical ventilation (41.6 vs 7.4 days, p < 0.001), length of ICU stay (44.3 vs 12.0 days, p < 0.001), and mortality (38 vs 3%, p < 0.001).
Conclusions: Mortality for patients who fail the first ICU attempt at delayed sternal closure is significantly higher than for those with successful sternal closure. Ventilatory pressures but not hemodynamic variables prior to DSC differed significantly between the two groups. First attempt at DSC was later in those who failed, suggesting that clinicians had a priori identified these patients as higher risk.
Article Information
vol. 130 no. Suppl 2 A19663
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Dept of Human Science, Georgetown Univ, Washington, DC
- 2Clinical Rsch Cntr, Boston Children’s Hosp, Boston, MA
- 3Cardiology, Boston Children’s Hosp, Boston, MA
- 4Cardiac Surgery, Boston Children’s Hosp, Boston, MA
Abstract 19928: Surgical Myectomy for Patients with Noonan Syndrome and Symptomatic Left Ventricular Outflow Tract Obstruction: A Case Control Series
Joseph T Poterucha, Patrick W O’Leary, Heidi M Connolly, Steve R Ommen, Michael J Ackerman, Hartzell V Schaff, Joseph A Dearani, Frank Cetta, Benjamin W Eidem
Circulation. 2014;130:A19928
Abstract
Introduction: Approximately 20-30% of patients (pts) with Noonan syndrome (NS) have asymmetric left ventricular hypertrophy (LVH) and LV outflow tract obstruction (LVOTO). The role of surgical myectomy in patients with NS and symptomatic LVOTO is unknown. The purpose of this study was to compare the clinical features and outcomes of pts with NS and LVOTO with age and gender matched patients with non-syndromic, obstructive hypertrophic cardiomyopathy (HCM) after myectomy.
Methods: Two cohorts were selected and retrospectively analyzed using Mayo Clinic databases from 1996-2014. Subjects included NS pts with LVH and LVOTO. Controls included 25 non-syndromic patients with obstructive HCM, matched to age, gender, and body surface area, who had myectomy during the same timeframe.
Results: 23 patients with NS and LVH were identified, of whom 12 underwent myectomy (8 males) for severe LVOTO [10 pediatric, 2 adult; 13 ± 10 yrs old (range 1 – 39)]. Pre and post-operative clinical and imaging data are shown in the Table. Ten septal and 2 combined septal/apical myectomies were performed in NS pts. Similar clinical and echocardiographic improvements were noted in both groups and most had only mild residual LVOTO (Table). There were no perioperative deaths. The majority showed improvement in NYHA class (7/8 NS versus 18/22 controls, 12 ± 13 months follow up). Decreased left atrial (LA) volume index was noted in HCM controls and was not found in NS pts (p < 0.05). Complications were rare: second myectomy was needed in 2 NS pts versus 1 control pt; heart block occurred in 1 control pt, and arrhythmias in 1 control pt. Only 1 late death in an NS pt occurred.
Conclusion: In NS pts with LVH and symptomatic LVOTO, myectomy reduces both gradient and NYHA class similar to results seen in pts with non-syndromic HCM. Post-operative morbidity was not increased. Residual gradients were slightly higher and greater LA dilation persisted in NS pts, but myectomy should not be avoided simply because of the presence of NS.
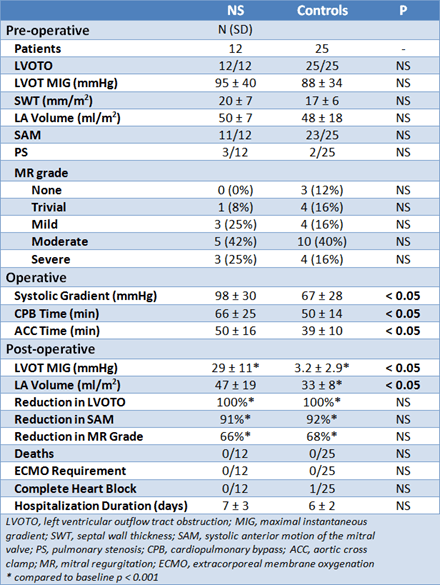
Article Information
vol. 130 no. Suppl 2 A19928
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Joseph T Poterucha1;
- Patrick W O’Leary1;
- Heidi M Connolly2;
- Steve R Ommen2;
- Michael J Ackerman1;
- Hartzell V Schaff3;
- Joseph A Dearani3;
- Frank Cetta1;
- Benjamin W Eidem1
- 1Pediatric Cardiology, Mayo Clinic College of Medicine, Rochester, MN
- 2Cardiovascular Diseases, Mayo Clinic College of Medicine, Rochester, MN
- 3Cardiac Surgery, Mayo Clinic College of Medicine, Rochester, MN
Abstract 18833: Annulus Preservation Strategy Improves Late Outcomes in Tetralogy of Fallot: An Anatomical Equivalency Study
Prisca Pondorfer, Tae-Jin Yun, Michael Cheung, David Ashburn, Cedric Manlhiot, Brian McCrindle, Luc Mertens, Lars Grosse-Wortmann, Andrew Redington, Glen Van Arsdell
Circulation. 2014;130:A18833
Abstract
Introduction: Late outcome of repaired TOF is driven by the impact of residual lesions. We shifted strategy from liberal transannular patch (TAP) use to aggressive valve and annulus preservation (AP) hypothesizing that for equivalent anatomy, AP would leave a mixed stenosis regurgitation lesion that would lead to a healthier right ventricle (RV).
Methods: Between 1996 and 2002, 185 children underwent TOF repair (median age 7.7 m). A regression equation for predicting annulus preservation, in the AP group, was derived from preoperative anatomic parameters and applied to all. Patients were identified (n=107) that could have had either AP or TAP on the basis of anatomical equivalency (subgroup validation with propensity matching) with 52 having a TAP and 55 having AP. These are the primary study group.
Results: Cardiac MRI at mean age 13.1±2.3 yrs (TAP n=28, AP n=23) showed AP was associated with significantly lower indexed RV end diastolic vol (AP: 120±29; TAP: 181±35 mL – Fig. 1), RV end systolic vol (AP: 57±23; TAP: 95±25 mL), RV stroke volume (AP: 64±15; TAP: 86±15 mL), MPA regurgitant fraction (AP: 28±11; TAP: 45±9 %), all p<0.0001, and LV mass (AP: 46±6; TAP: 54±8 gm/m2); p=0.001). Echo RVOT gradient was no different (AP 31 Vs TAP 25 mmHg (p=ns). MRI LVEF (AP: 57±4; TAP: 55±4 %; P=0.031) and RVEF (AP: 54±7; TAP: 48±6 %; p=0.004) was higher after AP. Freedom from surgical reintervention at 15 years was 89.3% (TAP) and 71.7% (AP , P=0.048) (Fig.2) with early reoperation for RVOTO predominating in AP and late pulmonary valve replacement most frequent in TAP. VO2 max for all AP vs TAP was higher in AP (p<0.05).
Conclusion: This is the first long-term follow-up study demonstrating that, for equivalent anatomy, an aggressive AP strategy leads to a lower reoperative incidence, less pulmonary insufficiency, smaller indexed RV volumes and better LV function as compared to a standard TAP. Surgical strategy directly impacts ventricular health and should be reflected in practice.
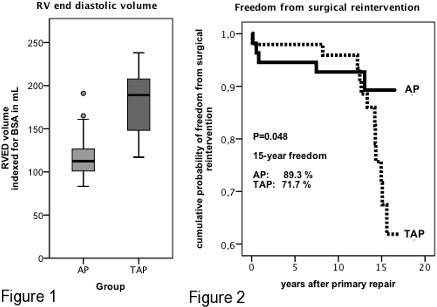
Article Information
vol. 130 no. Suppl 2 A18833
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Prisca Pondorfer;
- Tae-Jin Yun;
- Michael Cheung;
- David Ashburn;
- Cedric Manlhiot;
- Brian McCrindle;
- Luc Mertens;
- Lars Grosse-Wortmann;
- Andrew Redington;
- Glen Van Arsdell
- Cardiovascular Surgery, Sickkids Hosp, Toronto, Canada
Abstract 18211: Can Salvage Takedown of Cavopulmonary Connection Act as a Bridge to Subsequent Palliation?
Jiaquan Zhu, Devin Chetan, Arezou Saedi, John G Coles, Christopher A Caldarone, Glen S Van Arsdell, Osami Honjo
Circulation. 2014;130:A18211
Abstract
Objectives: Bidirectional cavopulmonary shunt (BCPS) and Fontan takedown are needed occasionally in failing single-ventricle palliation. We sought to analyze the outcomes of salvage takedown focusing on physiologic tolerance and subsequent clinical outcomes.
Methods: Twenty-four patients who underwent BCPS or Fontan takedown procedures from 1986 to 2013 were reviewed. Physiologic tolerance was defined as bridge to subsequent re-BCPS or re-Fontan, transplantation, and palliation suitable for future transplantation. Outcomes were compared using competing risk and Kaplan-Meier analysis.
Results: Nine (38%) patients underwent BCPS takedown [8 (89%) after 2000] and 15 (62%) patients underwent Fontan takedown [2 (13%) after 2000]. Nineteen (79%) patients had acute takedown (<1 month). The indications for BCPS takedown included hypoxia (n=7) and superior vena cava syndrome (n=2). Fontan takedown was due to low cardiac output (n=7), high Fontan pressure (n=5), cyanosis (n=1), persistent drainage (n=1), and protein losing enteropathy (n=1). Post-BCPS takedown physiology included 6 aortopulmonary shunt (APS) and 3 BCPS + APS, while post-Fontan takedown physiology included 12 BCPS + APS, 2 APS, and one 1.5 ventricle repair. The oxygen saturations at two weeks after BCPS takedown were significantly higher than before takedown (71.3±2.8% vs 41.7±14.6%, p=0.004). There were 10 (42%) in-hospital deaths and 3 (13%) late deaths. Overall, 11 (46%) patients were salvaged [3 (13%) re-BCPS/re-Fontan, 4 (17%) heart transplantation, 4 (17%) palliations suitable for later transplantation, Figure 1A]. All survivors were in NYHA class I-II except one who had failing BCPS+APS physiology. There was no significant difference in survival between BCPS and Fontan takedown (p=0.939, Figure 1B).
Conclusions: Cavopulmonary connection takedown procedures salvaged nearly half of patients despite substantial early mortality. The functional status of survivors is reasonable.
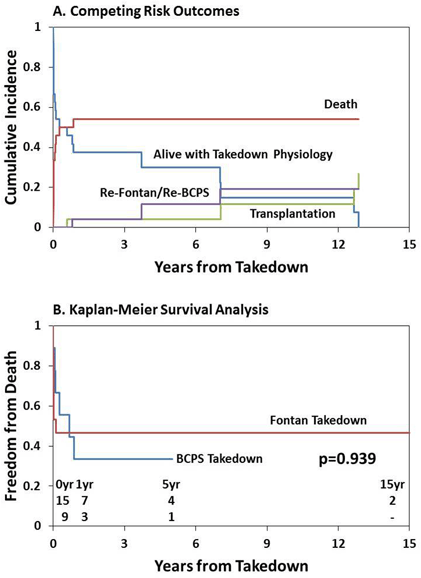
Article Information
vol. 130 no. Suppl 2 A18211
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Jiaquan Zhu;
- Devin Chetan;
- Arezou Saedi;
- John G Coles;
- Christopher A Caldarone;
- Glen S Van Arsdell;
- Osami Honjo
- Cardiovascular Surgery Div, The Hosp for Sick Children, Toronto, Canada
Abstract 16595: Airway Ciliary Dysfunction and Increased Postoperative Respiratory Complications in Broad Spectrum of Congenital Heart Disease Patients
Eileen Stewart, Maliha Zahid, Omar Khalifa, Ricardo Munoz, Brian Feingold, Peter Wearden, William Devine, Linda Leatherbury, Cecilia W Lo
Circulation. 2014;130:A16595
Abstract
Introduction: Congenital heart disease (CHD) patients with heterotaxy (HTX) have high prevalence of airway ciliary dysfunction (CD). This is associated with higher postoperative morbidity and more respiratory complications. As a high prevalence of airway CD is also observed in CHD patients without HTX, we hypothesize patients with broad spectrum of CHD may have increased postoperative morbidity associated with airway CD.
Methods: CHD patients <1 yr undergoing cardiac surgery were recruited. CD testing was conducted with two tests used for primary ciliary dyskinesia (PCD) diagnosis: nasal nitric oxide (nNO) was measured, typically low with PCD, and nasal scrape was obtained for ciliary motion (CM) analysis with videomicroscopy. Data on post-operative outcome parameters were collected.
Results: We recruited 55 CHD patients with 76 surgical events; 38% had airway CD (low nNO/ abnormal CM). Demographics and surgical complexity scores were similar between patients with or without CD. In CHD patients with CD, there was significant increase in continuous positive airway pressure ventilation (CPAP) (p = 0.008) and trend for more reintubation (11% non-CD vs. 27% CD patients) (Table 1). There was significant increase in beta-agonist use in CD patients (p = 0.006) and five-fold increase in respiratory medication use (Table 1). No difference was observed in length of stay, ventilator days, overall supportive ventilation (CPAP, high flow nasal cannula, nasal cannula), use of pulmonary vasodilators, chest tube days, effusion, or need for extracorporeal membrane oxygenation (ECMO).
Conclusions: CHD patients with airway CD have increased postoperative morbidity, including increased need for CPAP, more use of albuterol and respiratory medications, and more reintubations. These results suggest CHD patients may benefit from presurgical screening for airway CD, with those identified with CD given perioperative respiratory therapy to reduce postoperative morbidity.
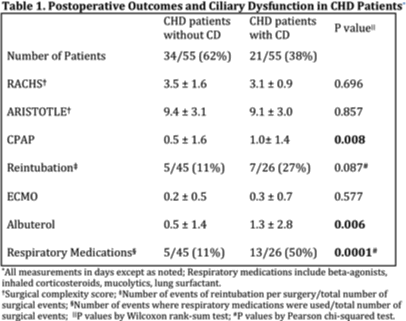
Article Information
vol. 130 no. Suppl 2 A16595
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Eileen Stewart1;
- Maliha Zahid2;
- Omar Khalifa2;
- Ricardo Munoz1;
- Brian Feingold1;
- Peter Wearden1;
- William Devine3;
- Linda Leatherbury4;
- Cecilia W Lo2
- 1Dept of Pediatrics, Children’s Hosp of Pittsburgh, Pittsburgh, PA
- 2Dept of Developmental Biology, Univ of Pittsburgh Sch of Medicine, Pittsburgh, PA
- 3Dept of Pathology, Univ of Pittsburgh Sch of Medicine, Pittsburgh, PA
- 4Dept of Pediatrics, Children’s National Med Cntr, Washington, DC
Abstract 16608: Building of a Right Ventricular Conduit in Native Outflow Tract in Preparation of Edwards Sapien Valve Implantation
- Giulia Gagliardi, Mara Pilati, Alessandra Cristofaletti, Salvatore Giannico, Aurelio Secinaro, Giacomo Pongiglione
Circulation. 2014;130:A16608
Abstract
Background: Percutaneous valve implantation (PPVI) is now considered feasible and safe especially in patients with conduit between the right ventricle and the pulmonary arteries. However there are still no clear indications on valvolization in native outflow tract (RVOT), traditionally considered at risk because of the dynamic characteristics of the RVOT
Aim: The aim of this study is to demonstrate the feasibility and safety of building a percutaneous conduit using a stent in the native RVOT before valve implant performing PPVI two months after stent implant, on the basis of previous animal studies, where endotelitation of the stent was shown to be reached within 60 days.
Methods: From October 2010 to April 2014, 19 pts, with tetralogy of Fallot, (mean age 15 ± 6 yrs) previously corrected by transannular patch technique, underwent a cardiac catheterization in order to build a stent-conduit in the native RVOT. After a complete right catheterization a balloon was inserted through a long sheath in the RVOT. During the inflation of the balloon a right ventricular angiography and a left coronary artery angiography are performed. In the presence of a complete occlusion of the RVOT and no coronary compression, a stent was deployed in the RVOT. Two months later an Edwards Sapien Pulmonary valve was implanted.
Results: Twenty two stents were implanted in 19 pts. The mean time of the procedures was 185 ± 89 min with a mean fluoroscopy time of 72 ± 33 min. Two main complications occurred: in 1 pt the stent partially occluded the right pulmonary artery and in a second pt the stent was dislocated in RVOT. In both cases the patients were treated surgically. In all other patients the procedure was successful and patients were discharged home two days later, on Aspirin therapy. The correct position of the stent was confirmed at the 1 month of follow-up by echocardiographic examination. Among the 19 pts , 152 underwent implantation of Edwards Sapien Pulmonary Valve, two months later. No complications occurred during the procedure and during the FU. Two patients are still waiting PPVI procedure
Conclusions: A two-step procedure approach increases the safety of PPVI with no impact on clinical conditions or complications
Article Information
vol. 130 no. Suppl 2 A16608
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- M. Giulia Gagliardi;
- Mara Pilati;
- Alessandra Cristofaletti;
- Salvatore Giannico;
- Aurelio Secinaro;
- Giacomo Pongiglione
- Med and Surgical Dept of Pediatric Cardiology, Ospedale Pediatrico Bambino Gesù, Rome, Italy
Abstract 15284: Characteristics and Determinants of Cerebral Circulation in Patients after Surgery for Congenital Heart Disease
Hirofumi Saiki, Seiko Kuwata, Clara Kurishima, Yoichi Iwamoto, Hirotaka Ishido, Satoshi Masutani, Hideaki Senzaki
Circulation. 2014;130:A15284
Abstract
Background: Neurodevelopmental delay and cerebral atrophy coupled with reduced blood flow are reportedly closely linked in patients with congenital heart diseases (CHD). However, the mechanisms to regulate cerebral flow in the same are unclear. Cardiac output and underlying cerebral arterial resistance that is constructed during infancy may affect blood perfusion. We tested our hypothesis that cerebral blood flow in patients with repaired CHD is affected by the original features of CHD.
Method: Ninety children including 50 patients with biventricular repaired CHD (ToF 27, TGA 15, CoA+VSD 8), 15 with a single ventricle (SV: Glenn7/Fontan8) and 25 with negligible shunts and otherwise normal stracture heart were enrolled. During cardiac catheterization, cardiac index (CI), blood pressure, central venous pressure were measured before and after inferior vena cava occlusion. Assuming that cerebral resistance (Rc) and lower body resistance (Ri) are constant during IVC occlusion, the resistance and blood distribution to the brain and lower body were calculated as a parallel resistance circuit. The impacts of hemodynamics on the cerebral circulation were analyzed.
Result: The cerebral portion of cardiac index (CIc) was negatively correlated with age (CIc=1.88-0.06*Age, p<.05) and positively correlated with the CI (CIc=0.26+0.35*CI, p<.0001). As expected, the cerebral resistance (Rc) increased significantly with age (p<.05), whereas the resistance ratio defined as the Rc/Ri was independent of age, body size, disease, and surgical history. Multivariate analysis revealed that the CI was the independent determinant of CIc, regardless of the underlying diseases (p<.01, β=0.27). Interestingly, after adjusting for age and CI, repaired CoA exhibited significantly higher Rs/Ri ratio (p<.01) and lower CIc (p<.05) than other groups, whereas SV patients showed significantly increased CIc, CIc/CI ratio and lower Rc compared to other groups (p<.05).
Conclusion: Apart from the strong governance of cerebral blood flow by cardiac output, cardiovascular property before anatomical repair and cavo-pulmonary connections have a significant impact on the cerebral circulation. Long-term effects of abnormal cerebral circulation warrants further studies.
Article Information
vol. 130 no. Suppl 2 A15284
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Hirofumi Saiki;
- Seiko Kuwata;
- Clara Kurishima;
- Yoichi Iwamoto;
- Hirotaka Ishido;
- Satoshi Masutani;
- Hideaki Senzaki
- Pediatric Cardiology, Saitama Med Cntr, Saitama Med Univ, Kawagoe, Saitama, Japan
Abstract 12664: Anomalous Aortic Origin of Coronary Arteries: A Single Center Experience
Assunta Fabozzo, Matthew DiOrio, Jane W Newburger, Andrew J Powell, Hua Liu, Francis Fynn-Thompson, Stephen P Sanders, Frank A Pigula, Pedro J del Nido, Meena Nathan
Circulation. 2014;130:A12664
Abstract
Background: Anomalous aortic origin of the coronary arteries (AAOCA) has an estimated prevalence of 0.1%-0.3%. Data on risks and benefits of surgery vs. observation to guide management decisions are limited.
Methods: In a single center, we included patients (pts) with AAOCA with right (R) and/or left (L) intramural (IM), interarterial IA) or intraconal (IC) course from 1996-2014. We excluded pts with benign CA anomalies or major structural heart disease.
Results: Among 154 pts, median age at diagnosis (dx) was 8.5 (range 0.1-50) yrs, median f/u was 1.92 (0.1-12.8) yrs, and 65% were male. The AAOCA course was IA in 116 (75%), IMin 52 (34%) and IC in 3 (2%). AAORCA was present in 126 (82%), of whom (51) 40% had repair. AAOLCA was present in 28 (18%), of whom 20 (71%) had repair; of the remaining 8, 2 had intraconal course; 2 were second opinions, 1 had Friederich’s ataxia, and 3 are awaiting surgery. In the surgical group, all had IA/IM CAs, and CA unroofing was performed in 90%, of whom 50% also had resuspension of the intercoronary commissure (table). Major perioperative complications occurred in 4 pts (6%: 1 ECMO, 1 mediastinitis, 2 early reoperation), 1 pt had late AoV repair. In the surgical group, no pts died; in the observed group, 2 pts with AAORCA (2.3%) died of severe non-cardiac comorbidities. Pts with surgery vs. observation were more likely to have AAOLCA (28% vs.10%, p=0.003), and, at time of dx, symptoms of chest pain/syncope (63% vs.13%, p<0.001), age >10 yrs (median 11 vs. 6 yrs, p<0.001), and exercise restriction (47% vs.13%; p<0.001). In multivariable modeling, surgical intervention was associated with chest pain or syncope (p<0.001) and older age (p=0.03).
Conclusion: In our center, all pts with AAOLCA with IM or IA course underwent surgery, but management of AAORCA was variable, and rare but serious complications occurred. Studies with long-term f/u are needed to develop evidence-based management guidelines for AAOCA patients.
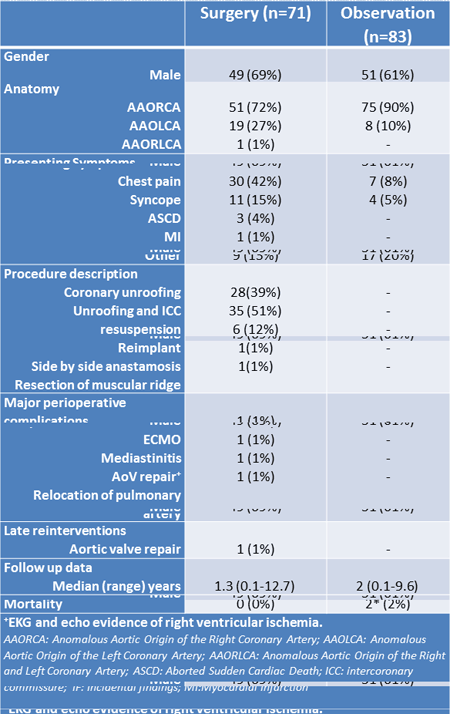
Article Information
vol. 130 no. Suppl 2 A12664
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Assunta Fabozzo1;
- Matthew DiOrio1;
- Jane W Newburger2;
- Andrew J Powell2;
- Hua Liu1;
- Francis Fynn-Thompson1;
- Stephen P Sanders1;
- Frank A Pigula1;
- Pedro J del Nido1;
- Meena Nathan1
- 1Cardiac Surgery, Boston Childrens Hosp, Harvard Univ, Boston, MA
- 2Cardiology, Boston Childrens Hosp, Harvard Univ, Boston, MA
Abstract 19783: Cerebral Oxygenation on Morning of Surgery Dependent on Time-to-Surgery in Infants with Critical Congenital Heart Disease
Jennifer M Lynch, Maryam Y Naim, Erin M Buckley, Madeline E Winters, Peter J Schwab, David R Busch, Ann L McCarthy, Tiffany S Ko, Rui Xiao, Susan C Nicolson, Lisa M Montenegro, Stephanie Fuller, J. William Gaynor, Thomas L Spray, Arjun G Yodh, Daniel J Licht
Circulation. 2014;130:A19783
Abstract
Introduction: Infants with critical congenital heart disease exhibit a high prevalence of hypoxic-ischemic white matter injury (WMI). Infants with transposition of the great arteries (TGA) have a greater risk for pre-operative injury, whereas infants with hypoplastic left heart syndrome (HLHS) have post-operative injury. Recent work has shown that increasing time from birth to surgery increases these risks for WMI in both cohorts. Understanding the changing preoperative cerebral physiology will help shed light on the differences in the timing of injury between diagnoses.
Methods: Term neonates with TGA or HLHS were recruited. Frequency domain diffuse optical spectroscopy and diffuse correlation spectroscopy were employed to noninvasively quantify cerebral oxygen saturation (ScO2) and cerebral blood flow (CBF) prior to surgery. Cerebral oxygen extraction fraction (OEF) was calculated from ScO2 and arterial oxygen saturation from arterial blood gases. Subjects were grouped by time-to-surgery, with one group undergoing surgery prior to day-of-life 4 and the other on day-of-life 4 or later.
Results: We studied 48 neonates diagnosed with either HLHS (N=30) or TGA (N=18). Anthropometric data were similar. Infants who went to surgery on day-of-life 4 or later had a significantly higher OEF (p < 0.01 (HLHS) and p = 0.02 (TGA)). There was no difference in CBF between groups in either diagnosis.
Conclusions: In patients with TGA, oxygen is limited only by decreased systemic oxygen levels, whereas in HLHS, oxygen delivery to the brain is limited by decreased blood flow in the aortic arch and, to a lesser extent, decreased systemic oxygenation. These results suggest that increasing cerebral oxygen demand preoperatively underlies the reported increased risk for WMI in infants with TGA and HLHS. The difference in timing of WMI between these diagnoses is more likely due to differences in cerebral oxygen delivery than metabolic demand.
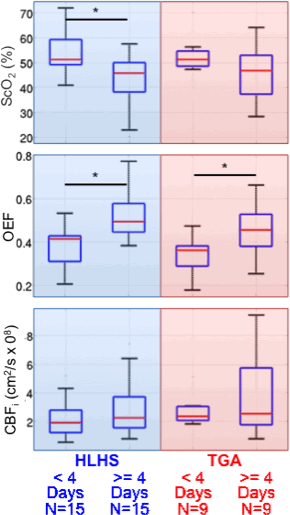
Article Information
vol. 130 no. Suppl 2 A19783
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Jennifer M Lynch1;
- Maryam Y Naim2;
- Erin M Buckley3;
- Madeline E Winters4;
- Peter J Schwab4;
- David R Busch4;
- Ann L McCarthy4;
- Tiffany S Ko5;
- Rui Xiao6;
- Susan C Nicolson7;
- Lisa M Montenegro7;
- Stephanie Fuller8;
- J. William Gaynor8;
- Thomas L Spray8;
- Arjun G Yodh1;
- Daniel J Licht4
- 1Dept of Physics, Univ of Pennsylvania, Philadelphia, PA
- 2Div of Critical Care Medicine, The Children’s Hosp of Philadelphia, Philadelphia, PA
- 3Optics Div, Athinoula A Martinos Cntr for Biomedical Imaging, Charlestown, MA
- 4Div of Neurology, The Children’s Hosp of Philadelphia, Philadelphia, PA
- 5Dept of Bionegineering, Univ of Pennsylvania, Philadelphia, PA
- 6Dept of Biostatistics, Univ of Pennsylvania, Philadelphia, PA
- 7Div of Cardiothoracic Anesthesia, The Children’s Hosp of Philadelphia, Philadelphia, PA
- 8Div of Cardiothoracic Surgery, The Children’s Hosp of Philadelphia, Philadelphia, PA
Abstract 19647: The Evolution of Cardiac Output and Cerebral Blood Flow in the Perinatal Transition in Hypoplastic Left Heart Syndrome: a Longitudinal Study
Lindsay Mills, Akiko Hirose, Santokh Dhillon, Etienne Fortin-Pellerin, Winnie Savard, Po-Yin Cheung, Deborah Fruitman, Nee S Khoo, Lisa K Hornberger
Circulation. 2014;130:A19647
Abstract
Background: Cardiovascular adaptation through the perinatal transition has not been previously explored in fetuses with hypoplastic left heart syndrome (HLHS). We sought to investigate the evolution of cardiac output in HLHS from late gestation through the perinatal period with comparison to healthy fetuses. As fetal HLHS has progressive middle cerebral arterial (MCA) Doppler changes suggestive of brain sparing, we further examined the impact of the transition on MCA blood flow. We hypothesized ductal patency with falling pulmonary vascular resistance after birth in HLHS could lead to cerebral steal.
Methods: We prospectively recruited 9 healthy and 11 pregnancies complicated by fetal HLHS. Echocardiograms were performed before birth at ≥37 weeks gestation and after birth at 4-6, 20-24, and 40-48 hours (all newborns), and 3-5 days (HLHS neonates only). Ventricular stroke volumes (SV) were determined using pulse Doppler and semilunar valve diameters, and combined cardiac output (CCO) was calculated (SV x heart rate (HR)). MCA pulsatility index (PI) was determined using the peak systolic, end diastolic and mean velocities.
Results: In late gestation, the CCO was similar between groups (HLHS mean 404 ± 91 vs. control 373 ± 96cc/kg/min). After birth, there was a trend towards higher CCO in HLHS at 4-6 and 20-24 hours (p= 0.06), which reached significance at 48 hours (587±155 vs controls 402±73 cc/kg/min, p=0.03). The CCO was highest in HLHS at 3-5 days (672 ± 117cc/kg/min, p<0.001 repeated measure ANOVA), due to higher HR and SV (for both versus controls, p<0.01). HLHS fetuses had significantly lower MCA PI than controls (1.2±0.1 vs 1.7±0.3, respectively, p<0.01); however, at 4-6 hours postnatally the MCA PI was higher in HLHS (1.8 ± 0.4 vs control 1.4±0.3, p= 0.04) and remained higher at 20-24 hours (p< 0.001) with higher systolic and lower end diastolic and mean velocities.
Conclusions: HLHS newborns significantly increase their cardiac output in the perinatal transition, particularly from 48 hours onward, through both increased HR and SV. While just prior to birth the MCA PI in HLHS suggests brain sparing, a reverse finding is observed after birth, which could reflect increased CCO and/or cerebral steal. This could be a period of cerebral vulnerability.
Article Information
vol. 130 no. Suppl 2 A19647
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Lindsay Mills1;
- Akiko Hirose1;
- Santokh Dhillon1;
- Etienne Fortin-Pellerin1;
- Winnie Savard1;
- Po-Yin Cheung1;
- Deborah Fruitman2;
- Nee S Khoo1;
- Lisa K Hornberger1
- 1Pediatric Cardiology, Univ of Alberta, Edmonton, Canada
- 2Pediatric Cardiology, Univ of Calgary, Calgary, Canada
Abstract 19033: Hyperoxia Reduces Oxygen Consumption in Children With Pulmonary Hypertension
Guo Long, Prashant Bobhate, Shreepal Jain, James Coe, Jennifer Rultledge, Ian Adatia
Circulation. 2014;130:A19033
Abstract
Background: High inspired oxygen concentration (FiO2) and inhaled nitric oxide (iNO), either alone or in combination, are administered to test pulmonary vasoreactivity in children. Oxygen consumption (VO2) cannot be measured accurately with conventional methods if the inspired FiO2 >0.85 and it is usually assumed that (VO2) does not change with hyperoxia or iNO.
Hypothesis: We hypothesised that an inspired FiO2 >0.85 would change VO2 compared with VO2 in room air (RA) and iNO administered with room air and could influence the accuracy of flow calculations using the direct Fick’s equation.
Methods: We reviewed retrospectively the cardiac catheterization data obtained between 2009-14 in children with pulmonary hypertension (PH) without cardiac shunts and cardiac output (CO) measured by thermodilution in 3 conditions of RA, with an FiO2 >0.85 and iNO in RA. VO2 was calculated using the Fick equation CO= VO2 / arterial-venous oxygen content difference. Dissolved O2 was included in the calculation.
Results: Data was available in 20 subjects (median age 8.5 years, range 1 -18, median weight 27 kg range, 8 – 95, BSA median 1m2, range 0.4 – 1.8). The median VO2 in RA was 147 (range 84-231) and decreased in hyperoxia to 134 (range 84-239) (p=0.03). The median percentage change in VO2 from RA to FiO2>0.85 was 10% (range + 24 to – 42%). VO2 was unchanged with iNO 139 (range 78-221) ml/min/m2 (p=0.3). The cardiac index remained unchanged in all 3 conditions (room air: 3.5±1.0, FiO2 >0.85: 3.5±1.0 and iNO: 3.6±1.0 L/min/m2).
Conclusion: VO2 decreased significantly during hyperoxia but not during iNO in children with PH. If VO2 is assumed to remain constant during hyperoxia errors maybe introduced if the direct Fick equation is used to calculate pulmonary and systemic blood flow.
Article Information
vol. 130 no. Suppl 2 A19033
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Pediatric Cardiology, Stollery Children’s Hosp, Univ of Alberta, Mazankowski Alberta Heart Institute, Edmonton, Canada
- 2Pediatric Cardiology, Stollery Children’s Hosp, Mazankowski Alberta Heart Institute, Univ of Alberta, Edmonton, Canada
Abstract 17907: Challenges of Anticoagulation during Cardiopulmonary Bypass in Children: Impact of Low Antithrombin Levels
Cedric Manlhiot, Colleen E Gruenwald, Helen M Holtby, Leonardo R Brandao, Glen S Van Arsdell, Brian W McCrindle
Circulation. 2014;130:A17907
Abstract
Introduction: Heparin-based anticoagulation in children is challenging as they have a high prevalence of heparin resistance. Antithrombin, one of the main natural anticoagulant inhibitors that potentiate heparin anticoagulation activity, might be a critical component of heparin response and anticoagulation effectiveness.
Objectives: Determine the impact of antithrombin levels on anticoagulation (heparin sensitivity and effectiveness) during cardiopulmonary bypass (CPB) in children <1 year old.
Methods: Secondary analysis of a randomized controlled trial of individualized vs weight based heparin management in 90 infants <1-year-old undergoing cardiac surgery. All analyses combined both patient groups and were used linear regression models.
Results: As expected, older patients had higher blood antithrombin levels (relative increase of 16±6% for patients >6 months old vs. those <6 month old, p=0.009). Response to heparin, reflected by change in anti-Xa levels from baseline after the initial heparin bolus (a laboratory measure of heparin sensitivity), was highest in patients with higher baseline antithrombin circulating levels (top tier: +0.94(0.16) U/ml per 100U/kg heparin vs. middle tier: +0.89(0.19) U/ml vs. lowest tier: +0.77(0.20) U/ml, p<0.001). Those patients with very low antithrombin levels during CPB (<0.35U/ml) had much greater heparin requirements (+165(30) U/kg/hr representing an increase of 243%, p<0.001) indicating greater heparin resistance. Insufficient anticoagulation on bypass (as measured by end of bypass anti-Xa level) was associated with higher clotting potential, reflected by higher plasma levels of thrombin-antithrombin complexes (+4.8(2.4) ng/ml per anti-Xa U/ml, p=0.05), and prothrombin activation fragment 1.2 (+85(32) pg/ml per anti-Xa U/ml, p=0.008). Moreover, lower circulating antithrombin level was associated with increased D-dimer levels after CPB (+53(25) ng/ml per 0.1 U/ml antithrombin, p=0.03).
Conclusions: Low antithrombin level is associated with resistance to heparin and decreased anticoagulation activity, ultimately leading to lower ability to suppress thrombin generation during CPB. Antithrombin supplementation may potentially individualize and improve anticoagulation.
Article Information
vol. 130 no. Suppl 2 A17907
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Cedric Manlhiot;
- Colleen E Gruenwald;
- Helen M Holtby;
- Leonardo R Brandao;
- Glen S Van Arsdell;
- Brian W McCrindle
- Labatt Family Heart Cntr, The Hosp for Sick Children, Toronto, Canada
Abstract 18067: Maladaptive Aortic Properties after the Norwood Procedure: an Angiographic Analysis of the Pediatric Heart Network Single Ventricle Reconstruction Trial
Sarah T Plummer, Christoph P Hornik, Hamilton Baker, Gregory A Fleming, Susan Foerster, Eric Ferguson, Andrew Glatz, Russel Hirsch, Jeffrey P Jacobs, Kyong-Jin Lee, Alan Lewis, Jennifer S Li, Mary H Martin, Diego Porras, Wolfgang A Radtke, John F Rhodes, Julie A Vincent, Jeffrey D Zampi, Kevin D Hill
Circulation. 2014;130:A18067
Abstract
Objectives: Aortic arch reconstruction in patients with single ventricle lesions may predispose to circulatory inefficiency and maladaptive physiology leading to increased myocardial workload. We sought to describe potentially maladaptive aortic anatomy and physiology, risk factors, and impact on right ventricle (RV) function in patients with single RV lesions after Norwood.
Methods: Pre-stage II angiograms for subjects from the Single Ventricle Reconstruction (SVR) Trial were compared to 50 single left ventricle controls with no prior arch reconstruction. Arch geometry was defined as normal, crenel (elongated), or gothic (angular). Aortic index (ascending /descending aorta diameter) and distensibility index (systolic aortic area – diastolic area) / (diastolic area x pulse pressure) were calculated. Univariable tests were used for comparisons, and Spearman’s rho was used for correlation.
Results: Interpretable angiograms were available for 326/389 (84%) SVR subjects. Table 1 summarizes findings. Age at catheterization was similar for SVR subjects and controls. SVR subjects more often demonstrated abnormally elongated (crenel) arch geometry with dilated ascending and transverse aortae tapering to a small isthmus. Distensibility was reduced in both the ascending and descending aorta. Risk factors for ascending aortic dilation (aortic index ≥ 2.5) included native aortic stenosis (OR = 2.2 [95% CI: 1.2, 3.8] vs. aortic atresia) and receipt of a RV-pulmonary artery shunt (OR = 2.6 [1.47, 4.5] vs. Blalock-Taussig shunt). There was no association between reduced distensibility and any demographic, anatomic, or reported surgical factors. Aortic dilation and reduced distensibility did not correlate with RV function at 14-month echocardiogram (p=0.5).
Conclusions: After Norwood single RV surgery, the reconstructed aorta demonstrates abnormal anatomy and physiology. Further study is needed to evaluate the longer-term impact of these findings.
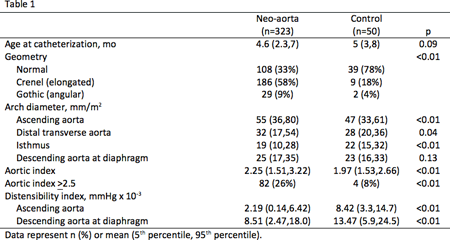
Article Information
vol. 130 no. Suppl 2 A18067
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Sarah T Plummer1;
- Christoph P Hornik1;
- Hamilton Baker2;
- Gregory A Fleming1;
- Susan Foerster3;
- Eric Ferguson4;
- Andrew Glatz5;
- Russel Hirsch6;
- Jeffrey P Jacobs7;
- Kyong-Jin Lee8;
- Alan Lewis9;
- Jennifer S Li1;
- Mary H Martin10;
- Diego Porras11;
- Wolfgang A Radtke12;
- John F Rhodes13;
- Julie A Vincent14;
- Jeffrey D Zampi15;
- Kevin D Hill1
- 1Pediatric Cardiology, Duke Univ Med Cntr, Durham, NC
- 2Pediatric Cardiology, Med Univ of South Carolina, Charleston, SC
- 3Pediatric Cardiology, Children’s Hosp of Wisconsin, Milwaukee, WI
- 4Pediatric Cardiology, Children’s Healthcare of Atlanta, Emory Univ Sch of Medicine, Atlanta, GA
- 5Pediatric Cardiology, Children’s Hosp of Philadelphia, Philadelphia, PA
- 6Pediatric Cardiology, Cincinnati Children’s Med Cntr, Cincinnati, OH
- 7Cardiovascular Surgery, John’s Hopkins Children’s Heart Surgery, All Children’s Hosp and Florida Hosp for Children, St Petersburg, FL
- 8Pediatric Cardiology, Hosp for Sick Children, Toronto, Canada
- 9Pediatric Cardiology, Children’s Hosp Los Angeles, Los Angeles, CA
- 10Pediatric Cardiology, Univ of Utah Sch of Medicine, Salt Lake City, UT
- 11Pediatric Cardiology, Children’s Hosp Boston, Boston, MA
- 12Pediatric Cardiology, Nemours Children’s Health System, Wilmington, DE
- 13Pediatric Cardiology, Miami Children’s Hosp, Miami, FL
- 14Pediatric Cardiology, Columbia Univ Med Cntr, New York, NY
- 15Pediatric Cardiology, Univ of Michigan Med Sch, Ann Arbor, MI
Abstract 17814: Prenatal Diagnosis is Associated with a Lower Rate of Pre-Operative Brain Injury in Newborns with Transposition of the Great Arteries
Shabnam Peyvandi, Veronica de Santiago, Patrick McQuillen
Circulation. 2014;130:A17814
Abstract
Objective: Literature suggests that prenatal diagnosis of critical congenital heart disease does not lead to improved mortality. However, it has been associated with better pre-operative conditions in single ventricle lesions and better neurodevelopmental outcome in transposition of the great arteries (TGA). We sought to describe the frequency of pre-operative brain injury in neonates with prenatal and postnatal diagnosis of TGA. We hypothesize that prenatally diagnosed neonates with TGA have a lower prevalence of pre-operative brain injury.
Methods: Term newborns with TGA were studied with brain magnetic resonance imaging preoperatively to determine brain injury severity based on the presence of white matter injury and/or stroke. Comparisons were made between prenatal and postnatal diagnosis. Other variables compared included estimated gestational age at birth (EGA), birth weight (BW), lowest oxygen saturation, balloon atrial septostomy (BAS), age at surgery, and hospital mortality. Analysis was performed with Fisher’s exact test or Student t-test. Further analysis will be performed including data from other centers and cardiac diagnoses.
Results: The presence of brain injury was significantly higher in postnatally diagnosed neonates than those prenatally diagnosed (Table). There were trends toward lower EGA, lower BW, higher oxygen saturation and younger age at surgery in the prenatal group. BAS did not differ by prenatal diagnosis. Mortality was low and the only two deaths were in the postnatal group.
Conclusions: Prenatally diagnosed neonates with TGA have a lower prevalence of preoperative brain injury as compared to those diagnosed postnatally. This finding is significant even when other known risk factors for brain injury did not differ between groups. Our data is consistent with prior literature demonstrating lower EGA and BW in prenatally diagnosed patients. The impact of these findings on neurodevelopmental outcomes will be studied.
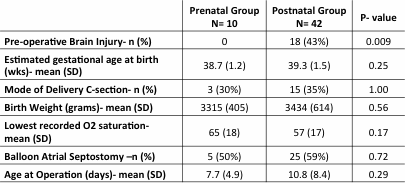
Article Information
vol. 130 no. Suppl 2 A17814
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Pediatric Cardiology, Univ of California San Francisco, San Francisco, CA
- 2Pediatrics, Univ of California San Francisco, San Francisco, CA
- 3Pediatrics and Neurology, Univ of California San Francisco, San Francisco, CA
Abstract 17849: Human Fetal Growth Model of Hypoplastic Left Heart Syndrome: Reduced Ventricular Growth Due to Decreased Preload
Sukriti Dewan, Adarsh Krishnamurthy, Roy C Kerckhoffs, Heather Sun, Jeffrey H Omens, Vishal Nigam, Andrew D McCulloch
Circulation. 2014;130:A17849
Abstract
Introduction: Hypoplastic Left Heart Syndrome (HLHS) is a congenital condition with an under-developed left ventricle (LV) that provides inadequate blood flow postnatally. Predicting LV size at birth using early stage fetal echocardiogram is a clinical challenge critical to the prognosis of HLHS. HLHS can develop during gestation due to altered biomechanical stimuli during fetal growth.
Hypothesis: We hypothesize that decreased preload in utero, from mitral stenosis/atresia, results in reduced LV growth in the fetal heart.
Methods: We developed a novel finite element (FE) model of the human fetal heart in which cardiac myocyte growth rates are a function of end-diastolic strain, which correlates with ventricular filling, to predict organ-level growth; and tested predictions with echocardiographic measurements in normal and hypoplastic fetal hearts. We computed LV filling as the difference between measured end-diastolic volume (EDV) and the mitral inflow; the FE model was made using human fetal geometry at 20wks.
Results: The strain-based fetal growth model with a normal 20wk preload (0.6ml) is able to replicate published measurements of mean LV EDV (0.9 to 8.3 ml; Fig.1) and LV dimensions (long-axis 18 to 35mm; short-axis 9 to 18mm) from mid-gestation to birth to within 10% r.m.s error. Decreasing preload (-25%) to 0.4ml at mid-gestation in the model, which emulates mitral stenosis in utero, a 50% reduction in LV EDV (3.5mL) and a 40% reduction in LV wall volume (2.4mL) is predicted at birth, similar to HLHS patients (Fig.1). In a blinded case study, using echocardiographic data (LV geometry and preload) from a HLHS patient at 30wks as the input, the model predicted a hypoplastic LV at birth, consistent with the patient diagnosis (Fig.1).
Conclusion: The human fetal growth model presented here is a significant step towards the development of a clinical tool that can be used to predict heart size in HLHS, based on LV filling at the end of the second trimester of pregnancy.
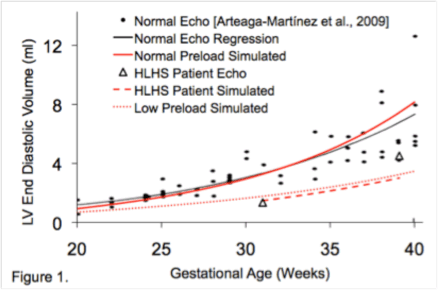
Article Information
vol. 130 no. Suppl 2 A17849
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Sukriti Dewan1;
- Adarsh Krishnamurthy1;
- Roy C Kerckhoffs1;
- Heather Sun2;
- Jeffrey H Omens1;
- Vishal Nigam3;
- Andrew D McCulloch1
- 1Bioengineering, Univ of California at San Diego, La Jolla, CA
- 2Pediatric Cardiology, Univ of California at San Diego; Rady’s Children Hosp, La Jolla, CA
- 3Pediatric cardiology, Univ of California at San Diego; Rady’s Children Hosp, La Jolla, CA
Abstract 16570: Respiratory Ciliary Motion Defect Predict Regional Brain Abnormalities and Increased Extra Axial CSF Fluid in Neonates With Complex Congenital Heart Disease
Ashok Panigrahy, Rafael Ceschin, Vince Lee, Nancy Beluk, Omar Khalifa, Giulio Zuccoli, Ricardo Munoz, Yuliya Domnina, Peter Wearden, Victor Morell, Maliha Zahid, Cecilia W Lo
Circulation. 2014;130:A16570
Abstract
Objectives: We hypothesized congenital heart disease (CHD) patients with respiratory ciliary motion (CM) defects may have increased prevalence of brain abnormalities.
Background: We recently showed CHD patients have a high prevalence of airway motile cilia dysfunction. In the brain, motile cilia in the ependyma mediate cerebrospinal fluid (CSF) flow required for neuronal cell migration/neurogenesis. Cilia are also found in the choroid plexus and are required for maintenance of neural progenitors. CHD patients with airway motile ciliary dysfunction may be at increased risk of neurocognitive abnormalities.
Methods and Results: 40 neonates with complex CHD underwent preoperative cranial US, volumetric MRI and respiratory ciliary motion (CM) assessment with high-speed videomicroscopy of nasal biopsies. CM was scored on a scale of 1 to 4, with 1 indicating normal CM, ranging to 4 for highly abnormal CM. Multivariate logistical regression of brain measurements with CM scores revealed significant correlation between respiratory CM defects and brain abnormalities (right panel, p<0.05), including hippocampal dysplasia/hypoplasia, increased extra-axial fluid, choroid plexus abnormalities (hypertrophy/nodularity) and corpus callosum dysplasia. We observed marginal correlations of CM defects with micrognathia, olfactory abnormalities and brainstem dysplasia. Correlation between volumetric increase in CSF and CM defects was observed using a neonatal brain segmentation template (r=0.64, p<0.007) (left panel).
Conclusions: Airway ciliary dysfunction is associated with structural brain abnormalities in neonates with complex CHD including: (1) subtle brain dysplasia in the olfactory bulb/hippocampus; (2) choroid plexus abnormalities and (3) increased extra-axial CSF fluid. These clinical translational studies suggest a potential unifying role for the cilia in contributing to both structural heart disease and brain abnormalities in CHD patients.
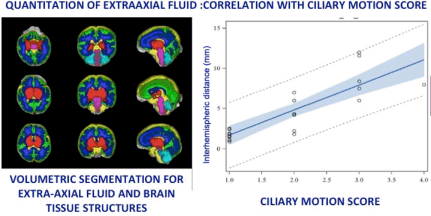
Article Information
vol. 130 no. Suppl 2 A16570
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Ashok Panigrahy1;
- Rafael Ceschin1;
- Vince Lee1;
- Nancy Beluk1;
- Omar Khalifa2;
- Giulio Zuccoli1;
- Ricardo Munoz1;
- Yuliya Domnina1;
- Peter Wearden1;
- Victor Morell1;
- Maliha Zahid2;
- Cecilia W Lo3
- 1Radiology, Children’s Hosp of Pittsburgh of UPMC, Pittsburgh, PA
- 2Cardiology, Children’s Hosp of Pittsburgh of UPMC, Pittsburgh, PA
- 3Developmental Biology, Children’s Hosp of Pittsburgh of UPMC, Pittsburgh, PA
Abstract 16148: Possible Implication of Oxidative Stress in Development of Coronary Vascular Lesions in Kawasaki Disease
Kenji Hamaoka, Tomoyo Yahata, Chinatsu Suzuki, Akiko Okamoto-Hamaoka, Ayako Yoshioka, Akihiro Nakamura, Kazuyuki Ikeda
Circulation. 2014;130:A16148
Abstract
Introduction: Kawasaki disease (KD) is an acute systemic pan-vasculitis prevalent in infants and consequently causes coronary arterial lesions (CALs). Furthermore, it has been reported that CALs in KD may increase the risk of early-onset and progression of arteriosclerosis. In this study, we evaluated the possible implication of oxidative stress (OS) in morbid state and developing vascular lesions by measuring the balance between oxidative stress and antioxidative activity.
Methods: Nineteen patients were enrolled to evaluate the dynamics of OS in the acute phase. All patients were treated with IVIG and aspirin. Thirteen patients responded well to IVIG and six did not respond well. Reactive oxygen metabolites (ROM) and biological antioxidant potential (BAP) were measured before (point A), two days (point B) and 1~2 weeks (point C) after IVIG administration, using FREE.
OS balance in the chronic-phase was examined in 42 late-phase KD patients (mean age; 16.5 years-old), who included 16 patients without CAL (group D), eight with transient CAL (group E), and 18 with persistent CALs (group F).
Results: In the acute phase of KD, OS was enhanced. ROMs in good response group significantly decreased between points A and B, and points B and C (p< 0.05 and p< 0.01, respectively). In poor response group, there was no significant difference in ROM between points A and B. BAP in good response group significantly increased between points A and C (p < 0.01), but not in poor response group. BAP at point A was significantly lower in the poor response group than the good response (p < 0.01).
No statistically significant differences in ROM and BAP were observed in groups D ~ F, but ROM was the lowest in group F. Potential antioxidative activity (BAP/dROMs) was significantly higher in group F compared with those in groups D and E (p< 0.05). In KD patients with CALs, ROM was the lowest in the combined therapy with antiplatelet medicine plus statin and/or warfarin, and AR was the highest.
Conclusions: In KD, OS was significantly enhanced in acute phase, and changes in ROM and BAP are clinically useful for evaluating and predicting the effects of IVIG therapy. Furthermore, it was suggested that combined treatment of statin or warfarin may increase potential antioxidant activity.
Article Information
vol. 130 no. Suppl 2 A16148
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Kenji Hamaoka;
- Tomoyo Yahata;
- Chinatsu Suzuki;
- Akiko Okamoto-Hamaoka;
- Ayako Yoshioka;
- Akihiro Nakamura;
- Kazuyuki Ikeda
- Pediatric Cardiology and Nephrology, Kyoto Prefectural Univ of Medicine, Kyoto, Japan
Abstract 15706: Survival in Children With Trisomy 21 Undergoing Single Ventricle Palliation
John Colquitt, Shaine A Morris, Susan Denfield, Charles D Fraser, William B Kyle
Circulation. 2014;130:A15706
Abstract
Background: Children with Trisomy 21 (T21) and single ventricle anatomy (SV) may have increased mortality compared to those without T21. Supporting data are limited. Our objectives were to describe survival in children with T21 and SV and determine risk factors associated with mortality.
Methods: We performed a single-center retrospective review. All patients born since 1980 with T21 and SV were included. We used Kaplan-Meier (KM) analysis with comparison by log rank to evaluate characteristics potentially associated with mortality in this cohort.
Results: Of 28 patients with T21 and SV, 26 (93%) had unbalanced atrioventricular septal defects with 13 (50%) being RV dominant. One patient had hypoplastic left heart syndrome and one had tricuspid atresia. 22 patients (79%) underwent pulmonary artery (PA) banding. Superior cavopulmonary connection (SCPC) was performed in 17 patients (61%) at a median age of 13 months (range 3 mo [[Unable to Display Character: –]] 13 y), with a banded PA left undivided in most. Three of these (18%) went on to total cavopulmonary connection (TCPC). Single-stage TCPC was performed in 2 other patients (7%). Median age at follow-up was 5 years (range 0.1 [[Unable to Display Character: –]] 33 y) overall and 7.5 years (range 1.5 [[Unable to Display Character: –]] 33 y) in survivors. One-year KM survival was 75% (95% CI 59-91%). Of 7 patients who died < 1 year old, only 1 had undergone SCPC. Two-year KM survival was 60% (95% CI 42-79%) with limited mortality beyond 2 years (Figure). Seventeen patients (61%) reached 5 years of age with 13 having SCPC by that age. Of 5 patients with TCPC, 4 are alive (age range 7 [[Unable to Display Character: –]] 14 y). Pulmonary vascular resistance (PVR) ≥ 3 WU*m2 in the first year of life was associated with increased mortality (p = 0.028) (Figure). No patient with PVR < 3 WU*m2 died. There was a trend toward association between ≥ moderate atrioventricular valve regurgitation and mortality (p = 0.071).
Conclusions: While T21 patients with SV remain a high-risk group, survival is excellent when PVR is < 3 WU*m2 in the first year of life with minimal attrition beyond 2 years of age.
Article Information
vol. 130 no. Suppl 2 A15706
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Pediatrics, Baylor College of Medicine/Texas Children’s Hosp, Houston, TX
- 2Pediatric Surgery, Baylor College of Medicine/Texas Children’s Hosp, Houston, TX
Abstract 13308: The Transcriptome of Kawasaki Disease Arteritis Reveals Activation of T Lymphocyte and Natural Killer Cell Function, Antigen Presentation, Immunoglobulin Production, and Interferon Response
Anne H Rowley, Kristine Wylie, Adam Pink, Rebecca Reindel, Susan Baker, Stanford Shulman, Jan Orenstein, Elizabeth Perlman, Mark Lingen, George Weinstock, Todd Wylie
Circulation. 2014;130:A13308
Abstract
Introduction: We recently published the new information that necrotizing arteritis, subacute/chronic vasculitis and luminal myofibroblastic proliferation are the three linked pathologic processes of Kawasaki Disease (KD) vasculopathy, which critically affects the coronary arteries (CA). The specific dysregulated immune pathways in KD CA have been unknown, and controversy exists as to whether immune dysregulation is primarily macrophage or T and B lymphocyte-driven.
Hypothesis: We hypothesized that the immune transcriptome of KD arteritis was primarily one of T and B lymphocyte rather than macrophage activation.
Methods: RNA was isolated from paraffin-embedded CA tissues. RNA samples passing quality control assays were subjected to ribosomal RNA subtraction, and Illumina HiSeq 2000 RNA sequencing (101 nt paired-end reads, 40-100million reads/sample) was performed. Reads were aligned to the human genome, and Cuffdiff used to test for differential gene expression in CA tissues from 8 KD children (median age=7 mo, median 4 wks from onset) compared to 7 controls who had normal CA pathology (median age=5 mo). Pathways analysis was performed using Ingenuity iReport™.
Results: 1057 differentially expressed transcripts were identified with >1.5-fold change and q-value <0.05. The most significantly upregulated pathways included T lymphocyte, dendritic cell and natural killer cell signaling, antigen presentation, and toll-like receptor (TLR) signaling (p values 1e-18 to 5e-05). Immunoglobulin A, G, and M genes and at least 50 type I interferon-stimulated genes were significantly upregulated. Tumor necrosis factor α (TNFα) receptor and transforming growth factor β signaling pathways were not significantly altered (p-value >0.1). 50 immune response genes encoding secreted proteins were upregulated and are candidate biomarkers of KD arteritis.
Conclusions: The host transcriptional response in target CA tissues in KD demonstrates a prominent innate and acquired immune response to antigen. Genes related to T and B lymphocyte activation were upregulated but macrophage responses were not prominent. This first report of the CA transcriptome of KD could lead to identification of new therapeutic targets and biomarkers for KD arteriopathy.
Article Information
vol. 130 no. Suppl 2 A13308
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Anne H Rowley1;
- Kristine Wylie2;
- Adam Pink1;
- Rebecca Reindel1;
- Susan Baker3;
- Stanford Shulman4;
- Jan Orenstein5;
- Elizabeth Perlman6;
- Mark Lingen7;
- George Weinstock8;
- Todd Wylie8
- 1Pediatrics, Northwestern Univ Feinberg Sch of Medicine, Chicago, IL
- 2Pediatrics, Washington Univ, St Louis, MO
- 3Microbiology and Immunology, Loyola Univ Stritch Sch of Medicine, Maywood, IL
- 4Pediatrics, Ann & Robert H Lurie Children’s Hosp of Chicago, Chicago, IL
- 5Pathology, George Washington Univ Sch of Medicine, Washington, DC
- 6Pathology, Ann & Robert H Lurie Children’s Hosp of Chicago, Chicago, IL
- 7Pathology, Univ of Chicago Pritzker Sch of Medicine, Chicago, IL
- 8The Genome Institute, Washington Univ, St Louis, MO
Abstract 16397: Correlates of Successfully Achieving Statin Therapy Targets in Pediatric Patients
Michael Mendelson, Todd Regh, Heather Harker, Nicole Palumbo, James Nevin, Phillip K Johnson, Suzanne Griggs, Meera Boghani, Nirav Desai, Elizabeth Yellen, Lucy Buckley, Annette Baker, Mathew Gillman, Justin Zachariah, Dionne Graham, Sarah D de Ferranti
Circulation. 2014;130:A16397
Abstract
Introduction: Targeting LDL-C goals for statin therapy is currently recommended for children and adolescents in NHLBI pediatric guidelines; however, there is limited information about clinical factors, especially modifiable ones, that impact successful achievement of LDL-C goals.
Methods: We analyzed data prospectively collected as part of a quality improvement initiative, SCAMP® (Standardized Clinical Assessment and Management Program), in the Preventive Cardiology Clinic at Boston Children’s Hospital, Boston, MA. We included patients initiated on statin therapy from September 2010 to March 2014. Achieving LDL target was defined as an LDL-C < 130 mg/dL or an LDL-C < 100mg/dL with a high level risk factor (i.e. diabetes, etc.). The impact of baseline CVD risk factors were assessed in Cox proportional hazards models.
Results: Among the 1521 pediatric patients and 3813 clinical encounters that occurred over the 3.5 year period, 116 patients were defined as initiating statin therapy. Complete data was available on 79 patients (mean [SD] age 14 [4] years; 47% female; median [IQR] LDL-C 215 [79] mg/dL). The probability of achieving LDL-C goal within 365 and 500 days of starting a statin was 0.55 (95% CI 0.44-0.68) and 0.67 (0.55-0.79), respectively; Figure 1. In univariate analyses, female sex (p=0.01) and lower baseline LDL-C (p=0.04) was associated with more rapid achievement of LDL-C goals, but not age (p=0.8), BMI (p=0.2), exercise (p=0.6), nutritional factors (p=0.3), screen time (p=0.3), cigarette smoking (p=0.9), or family history of early CVD (p=0.3).
Conclusions: The majority of children reached LDL-C treatment goals with statin therapy. Baseline modifiable lifestyle CVD risk factors such as diet and exercise were not associated with achieving LDL-C goal on statin therapy in children and adolescents. Increased support and monitoring may be needed, particularly for males, in order to achieve therapy goals.
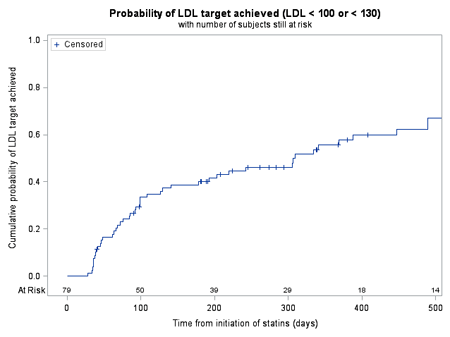
Article Information
vol. 130 no. Suppl 2 A16397
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Michael Mendelson1;
- Todd Regh1;
- Heather Harker1;
- Nicole Palumbo1;
- James Nevin1;
- Phillip K Johnson1;
- Suzanne Griggs1;
- Meera Boghani1;
- Nirav Desai1;
- Elizabeth Yellen1;
- Lucy Buckley1;
- Annette Baker1;
- Mathew Gillman2;
- Justin Zachariah1;
- Dionne Graham1;
- Sarah D de Ferranti1
- 1Cardiology, Boston Children’s Hosp, Boston, MA
- 2Dept of Population Medicine, Harvard Pilgrim Healthcare, Boston, MA
Abstract 15915: Gender Matters in the Risk for Sudden Death in Young Competitive Athletes
Aneesha Ahluwalia, Tammy S Haas, Ross F Garberich, Barry J Maron
Circulation. 2014;130:A15915
Abstract
Background: Sudden deaths in young competitive athletes are highly visible and tragic events, which receive much public visibility. However, little is known regarding the specific impact of gender on these events.
Methods and Results: The U.S. National Registry of Sudden Death in Athletes was accessed to identify the causes, frequency and epidemiology of sudden deaths in U.S. competitive athletes by gender. A total of 2,408 deaths (2155 male; 253 female) were identified in athletes engaged in 41 diverse organized sports from 1980 to 2011. Most sudden deaths occurred with physical exertion during competition/training (66%), and were predominantly due to cardiovascular disease (54%). In high school and college athletes, accounting for participation rates, the ratio of male to female deaths was 5.4:1. Males were somewhat older than females (19±6 vs. 17.8±6; P=0.003), and were more commonly black (30% vs. 17%; P<0.001). Of the male athletes, the highest rate of cardiovascular sudden death was in blacks (65% vs. 49% in whites; P<0.001). The most frequent sports were football (n=725) and basketball (n=437) in males, and basketball (n=47) and cross country/track (n=41) in females. Most common cardiovascular cause of death was hypertrophic cardiomyopathy (HCM), which accounted for a greater proportion of confirmed cardiovascular deaths in males (47%) vs. females (14%; P<0.001); a 19:1 male:female ratio in high school and college athletes. In contrast, females experienced greater frequency of death due to congenital coronary artery anomalies (30% vs. 14%; P<0.001), arrhythmogenic right ventricular dysplasia (12% vs. 4%; P=0.018), and long-QT syndrome (7% vs. 1%; P<0.001). Blunt trauma, including commotio cordis, occurred more frequently in males (24% vs. 15% in females; P=0.001).
Conclusions: Female athletes have a 5-fold lower risk for sudden death, compared to males, and were almost 20-times less likely to die of HCM. Black males had the highest rate of sudden death due to cardiovascular disease, with HCM most common.
Article Information
vol. 130 no. Suppl 2 A15915
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Aneesha Ahluwalia;
- Tammy S Haas;
- Ross F Garberich;
- Barry J Maron
- Hypertrophic Cardiomyopathy Cntr, MInneapolis Heart Inst Foundation, Minneapolis, MN
Abstract 14841: Energy Drink Exposures in the National Poison Data System: Epidemiology and Clinical Effects
Steven A Seifert, Kristopher L Arheart, Vivian I Franco, Alvin C Bronstein, Stacy D Fisher, Brandon J Warrick, Sara M Seifert, Steven E Lipshultz
Circulation. 2014;130:A14841
Abstract
Introduction: Energy drinks (ED) are beverages whose stimulant properties are likely linked to their caffeine content, including pharmaceutical caffeine and, in some cases, additional caffeine from natural sources, other additives and, prior to the FDA ban, ethanol. Of particular concern are exposures in children, especially those with underlying cardiac or neurological conditions.
Methods: This is a retrospective analysis of human exposures to single-substance ED formulations in the National Poison Data System of the American Association of Poison Control Centers from 10/1/10 – 9/30/13.
Results: There were 10,610 cases of ED exposure, with 5,156 being single-substance exposures with identifiable contents. Over 40% involved unintentional exposures by children < 6 years old, which was constant over the study period. There were, however, increases in unintentional ED exposures (p=0.002) and no-effect outcomes (p=0.015) over time. Outcome severity was increased with exposure to products containing caffeine from multiple sources v. pharmaceutical caffeine only (23% v. 15% “Moderate” or “Major” outcomes, p<0.001). The most prevalent organ system effects in non-ethanol-containing ED were neurologic in 20%, GI in 15% and cardiovascular in 12%. Exposures with “Major” outcomes had cardiovascular effects in 57% (including rhythm and conduction abnormalities), and neurologic effects in 55% (seizures, including status epilepticus). Ethanol as an additive was also associated with increased outcome severity v. non-ethanol-containing ED (42% v. 19% “Moderate” or “Major” outcomes, p<0.001). There has been a continued decline in reports of exposure to combination ED-ethanol products since the FDA ban.
Conclusions: ED exposures reported to poison centers demonstrate a sizable proportion of pediatric cases, some with serious cardiac and neurological effects. Demographic changes over time may be related to public and professional educational efforts. Observed clinical effects are consistent with known effects of caffeine. Regulatory action has been followed by a decrease in poison center reports of ethanol-containing ED. We recommend improved labeling of caffeine content and continued efforts to decrease pediatric exposures to these products.
Article Information
vol. 130 no. Suppl 2 A14841
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Steven A Seifert1;
- Kristopher L Arheart2;
- Vivian I Franco3;
- Alvin C Bronstein4;
- Stacy D Fisher5;
- Brandon J Warrick1;
- Sara M Seifert6;
- Steven E Lipshultz7
- 1Emergency Medicine, Univ of New Mexico / New Mexico Poison Cntr, Albuquerque, NM
- 2Public Health, Univ of Miami Miller Sch of Medicine, Miami, FL
- 3Pediatrics, Wayne State Univ Sch of Medicine and Children’s Hosp of Michigan, Detroit, MI
- 4Emergency Medicine, Rocky Mountain Poison and Drug Cntr / Univ of Colorado Sch of Medicine, Denver, CO
- 5Internal Medicine / Pediatrics, Univ of Maryland Sch of Medicine, Baltimore, MD
- 6Gynecology & Obstetrics, Johns Hopkins Med Cntr, Baltimore, MD
- 7Pediatrics, Wayne State Univ Sch of Medicine, Detroit, MI
Abstract 11803: Factors Associated With Delayed Diagnosis of Critical Congenital Heart Defects in Texas
Unnati Doshi, Syed S Hashmi, Lisa Marengo, Surabhi Kaul, Mousumi Moulik
Circulation. 2014;130:A11803
Abstract
Purpose: Critical congenital heart defects (CCHD) require surgical and/or transcatheter intervention in infancy. They may or may not have ductal dependent circulation. Timely diagnosis before ductal closure is important to reduce morbidity and mortality. Our goal was to identify factors associated with delayed diagnosis of CCHD.
Methods: This is a retrospective, population-based, observational study of liveborn infants in Texas born from 2000 – 2009 identified using the Texas Birth Defects Registry (TBDR). Inclusion required a definitive diagnosis of one of 13 selected CCHDs identified by diagnostic code and information on one of three “diagnostic” procedures: prenatal or postnatal echocardiogram or autopsy. Cases with aortic or pulmonary stenosis, coarctation of aorta, tricuspid stenosis or Ebsteins anomaly were included only if they underwent a cardiac surgical or catheterization procedure within 6 weeks of life. Stillborn infants or that died within three days of life (DOL) without evidence of an echocardiogram procedure were excluded. Date of diagnosis was defined as date of first echocardiogram. “Delayed diagnosis” was defined as any date of diagnosis after DOL3. Eligible infants with autopsy as the only “diagnostic” procedure were also classified as “delayed diagnosis”. Logistic regression models were created to identify factors that influence diagnosis delay.
Results: A total of 5895 CCHD cases were identified. Diagnosis was delayed in 25% of cases. The CCHDs with highest rates of delayed diagnosis were critical coarctation (34%) and TAPVR (32%). In multivariate analysis, delayed diagnosis was more likely to be associated with single CCHD [adjusted odds ratio (aOR) 1.9; 95% CI 1.5-2.4], low maternal education [aOR1.7; 95% CI 1.2-2.3], prematurity <32wks [aOR 1.6; 95% CI 1.21-2.26], and rural maternal residence [aOR 1.5; 95% CI 1.22-1.94]. Presence of noncardiac defects decreased delayed diagnosis only for a few CCHD phenotypes.
Conclusions: A quarter of all births with CCHD have a delayed diagnosis in Texas. Maternal education and residence are associated with the delay. Future studies will have to investigate each CCHD phenotype for specific risk factors.
Article Information
vol. 130 no. Suppl 2 A11803
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Dept of Pediatric Cardiology, Univ of Texas – Houston Med Sch, Houston, TX
- 2Dept of Pediatrics, Univ of Texas – Houston Med Sch, Houston, TX
- 3Birth Defects Epidemiology and Surveillance Branch, Texas Dept of State Health Services, Austin, TX
- 4Sch of Public Health, Univ of Texas Sch of Public Health, Houston, TX
Abstract 19711: Novel Insights and Reference Intervals of Soluble ST2 in a Pediatric Population Free of Heart Failure
Jeffrey W Meeusen, Jonathan N Johnson, Allan S Jaffe, Leslie J Donato, John L Jefferies, Aarti Dalal, Kevin O Maher, Amy K Saenger
Circulation. 2014;130:A19711
Abstract
Introduction: Soluble ST2 is a novel prognostic biomarker in adults with chronic heart failure. The utility of ST2 in pediatric patients with heart failure and other cardiovascular co-morbidities is limited due to lack of normal interpretive values. We sought to determine the reference intervals of ST2 in a healthy pediatric population.
Methods: Sera from 240 healthy children were identified from an institutional pediatric biobank at the Mayo Clinic (Rochester, MN). 40 male and 40 female subjects within three age groups (2-6 years, 7-12 years and 13-17 years) were included in the study and those with a diagnosis of anemia, autoimmune disease, hematologic disease/bleeding, circulatory/heart failure, kidney or liver disease, malignancy, malnutrition, diabetes, or pregnancy were excluded. ST2 was measured using a novel high-sensitivity sandwich immunoassay (Presage ST2 assay; Critical Diagnostics, San Diego, CA, USA). Parametric analysis established the 95th percentile reference interval between genders and age groups.
Results: ST2 was not significantly associated with age, gender or body mass index (BMI). The median ST2 across the entire cohort was 21 ng/mL (range: 6 – 122 ng/mL). The central 95th percentile was 8 – 64 ng/mL; cut-points for the 90th and 95th percentile were 38 and 48 ng/mL, respectively. Four outliers were excluded due to an ST2 greater than 2 times the interquartile range from the next nearest value. The final central 95th percentile reference interval was 9-50 ng/mL with cut-points for the 90th and 95th percentile at 37 and 43 ng/mL, respectively (median remained 21 ng/mL). Association between ST2 and other diagnoses or medications was not statistically significant.
Conclusions: This study determined a normal reference interval for ST2 in pediatric patients without heart failure which aids in effective interpretation of ST2 in a diseased pediatric cohort. The median and upper-limit of normal in pediatric ST2 (21 and 50 ng/mL) were higher than that for adults (19 and 35 ng/mL). Further studies in pediatric cohorts, both normal and diseased, are warranted to evaluate disease-specific trends and clinical decision limits in pediatric heart failure.
Article Information
vol. 130 no. Suppl 2 A19711
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Jeffrey W Meeusen1;
- Jonathan N Johnson2;
- Allan S Jaffe3;
- Leslie J Donato4;
- John L Jefferies5;
- Aarti Dalal6;
- Kevin O Maher6;
- Amy K Saenger7
- 1Clinical Chemistry, Mayo Clinic, Rochester, MN
- 2Pediatric Cardiology, Mayo Clinic, Rochester, MN
- 3Cardiovascular Diseases, Mayo Clinic, Rochester, MN
- 4Laboratory Medicine, Mayo Clinic, Rochester, MN
- 5Pediatric Cardiology, Cincinnati Children’s Hosp, Cincinnati, OH
- 6Pediatric Cardiology, Emory Univ, Atlanta, GA
- 7Med and Scientific Affairs, Roche Diagnostics, Indianapolis, IN
Abstract 19370: Myocardial Extracellular Volume is Elevated in Human Duchenne Muscular Dystrophy
Jonathan H Soslow, Stephen M Damon, Bruce M Damon, David A Parra, W B Burnette, Douglas B Sawyer, C M Stein, Larry W Markham
Circulation. 2014;130:A19370
Abstract
Introduction: Duchenne muscular dystrophy (DMD) leads to cardiomyopathy (CM) with variable severity and age of onset. Predicting early CM would alter therapeutic approaches and improve morbidity and mortality. Extracellular volume (ECV) calculated with cardiac MRI (CMR) quantifies extracellular matrix expansion, including myocardial fibrosis, and has never been reported in human DMD. We hypothesized that subjects with DMD would have abnormal ECV and that these values would correlate with other markers of LV function.
Methods: 27 DMD subjects were prospectively studied. CMR included LVEF, late gadolinium enhancement (LGE), circumferential strain (εcc), and modified Look-Locker (MOLLI) sequences to calculate ECV maps (in-house software using Matlab). ECV calculated for each segment in the short axis at mid-ventricle and compared with LVEF and εcc using linear regression. Normal values taken from unmatched cohort of healthy male adults with mean ECV of 0.25 ± 0.015 (range 0.23-0.28).
Results: Imaging was adequate to calculate ECV maps in 20 DMD subjects. Mean age in years was 14. Mean LVEF was 52% and mean global εcc was -14.6%; 11 subjects had LVEF < 55% and 6 had negative LGE. Mean ECV was 0.33 ± 0.05 (0.25-0.45); there was significant intersubject and intersegment variability (Figure 1). When compared with highest mean control ECV (0.28), only one subject had a normal ECV in every segment. In subjects with LVEF ≥ 55%, the mean ECV was 0.32 ± 0.07 (0.25-0.45). In subjects with negative LGE, the mean ECV was 0.28 ± 0.04 (0.25-0.36). The ECV of the inferolateral and anterolateral segments correlated with LVEF (p=0.025 and p<0.001) and the inferolateral ECV correlated with mean εcc (p=0.025).
Conclusions: In this cohort, 19/20 DMD subjects have elevated segmental ECV, even with normal LVEF and negative LGE. Segmental ECV correlates with both LVEF and mean εcc. ECV may be a more subtle biomarker of early myocardial disease than standard measures such as LVEF and LGE in human DMD.
Article Information
vol. 130 no. Suppl 2 A19370
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Jonathan H Soslow1;
- Stephen M Damon2;
- Bruce M Damon3;
- David A Parra1;
- W B Burnette4;
- Douglas B Sawyer5;
- C M Stein6;
- Larry W Markham7
- 1Pediatrics, Div of Pediatric Cardiology, Vanderbilt Univ Med Cntr, Nashville, TN
- 2Neurology, Vanderbilt Univ Med Cntr, Nashville, TN
- 3Radiology and Radiological Sciences, Molecular Physics and Biophysics, and Biomedical Engineering, Vanderbilt Univ Med Cntr, Nashville, TN
- 4Pediatrics, Div of Neurology, Vanderbilt Univ Med Cntr, Nashville, TN
- 5Medicine, Div of Cardiovascular Medicine, Vanderbilt Univ Med Cntr, Nashville, TN
- 6Medicine, Pharmacology, Vanderbilt Univ Med Cntr, Nashville, TN
- 7Pediatrics, Div of Pediatric Cardiology; Medicine, Div of Cardiovascular Medicine, Vanderbilt Univ Med Cntr, Nashville, TN
Abstract 15668: Longitudinal Changes in High Sensitivity Troponin T and Pro-B-Type Natriuretic Peptide Over Two Post-Transplant Years in Rejection-Free Pediatric Heart Recipients
Fatima I Lunze, Tajinder P Singh, Nader Rifai, Kimberlee Gauvreau, Ryan Narciso, Kevin P Daly, Leslie B Smoot, Christina VanderPluym, Elizabeth D Blume, Steven D Colan
Circulation. 2014;130:A15668
Abstract
Purpose: High sensitivity troponin T (hs-TnT) and pro-B-type natriuretic peptide (pro-BNP) assays may be of value for rejection-surveillance in heart transplant (HT) recipients, a use that requires a better understanding of their behavior in rejection-free patients.
Hypothesis: The serum concentrations of hs-TnT, pro-BNP and four other biomarkers will be elevated early post-HT and decline over time in rejection-free pediatric HT recipients.
Methods: We measured serial biomarker concentrations in 24 children who received HT (median age at HT 6.6 yrs, range 0.5-16) at our institution between March 2013 and January 2014 and remained rejection-free by biopsy. All blood samples were prospectively collected at the time of biopsy. Patients with rejection (defined as ISHLT cellular Grade ≥2R or antibody mediated rejection) were excluded. Hs-TnT, pro-BNP, hs-CRP, ST2 (an interleukin 1 receptor inhibitor), galectin-3, and vascular endothelial growth factor (VEGF) were measured from frozen stored serum samples.
Results: We obtained 81 frozen serum samples (median 3 samples per patient, range 2- 7). There was a significant decline in hs-TnT, pro-BNP and hs-CRP levels within first 6 months after HT (all P ≤ 0.005) but not in ST2, Galectin-3 and VEGF levels. The serum concentrations of biomarker values in median (25th, 75th percentiles) by time since HT are shown in Figure 1. Higher hs-TnT and ST-2 concentrations were associated with younger donor age (r= -0.65, p= 0.06 and r= -0.66, p= 0.05) and longer ischemic time (r= 0.67, p= 0.05 and r= 0.75, p= 0.02).
Conclusion: In this prospective study, we found marked elevation of serum concentrations of hs-TnT, pro-BNP and hs-CRP early after HT. Levels declined over time in pediatric HT recipients who remained rejection-free. Adjusting for time since HT may improve the diagnostic utility of these biomarkers for non-rejection surveillance protocols.
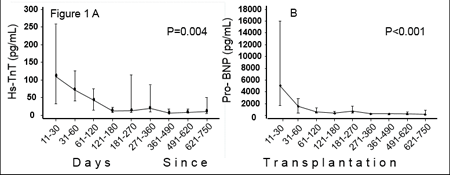
Article Information
vol. 130 no. Suppl 2 A15668
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Fatima I Lunze;
- Tajinder P Singh;
- Nader Rifai;
- Kimberlee Gauvreau;
- Ryan Narciso;
- Kevin P Daly;
- Leslie B Smoot;
- Christina VanderPluym;
- Elizabeth D Blume;
- Steven D Colan
- Dept of Cardiology, Boston Children’s Hosp, Boston, MA
Abstract 11572: Hyperuricemia Reflects Heart Failure Severity and Predicts Morbidity and Mortality in Patients After the Fontan Operation
Hideo Ohuchi, Yosuke Hayama, Jun Negishi, Kanae Noritake, Osamu Sasaki, Nobuyuki Tsujii, Toru Iwasa, Heima Sakaguchi, Aya Miyazaki, Osamu Yamada
Circulation. 2014;130:A11572
Abstract
Background: Serum uric acid level (UA, mg/dl) not only associates with pathophysiology in adult patients with chronic heart failure but also predicts the cardiac events. However, the clinical significance of UA remains unclear in patients after the Fontan operation.
Purpose: To clarify clinical significance of hyperuricemia (HUA, UA≥7.0) in Fontan patients.
Methods and Results: We prospectively measured UA in consecutive 306 Fontan patients (16±8 years) and compared the results with the clinical variables, including hemodynamics, exercise capacity, plasma levels of norepinephrine (NE), brain natriuretic peptide (BNP), and unscheduled hospitalization (USH), including all-cause mortality. The UA was 5.8±1.7 and 70 (23%) patients showed HUA. On multivariate model, high levels of plasma creatinine and NE, use of diuretics, low cardiac index, hypoxia, and hyponatrenia independently associated with HUA (p<0.05-0.001), however BNP level did not correlate with UA (p=0.35). During a follow-up of 49±27 months, 69 USH, including 16 deaths, occurred. In addition to BNP levels, HUA independently predicted USH (hazard ratio [HR]: 2.9, 95% confidence interval [CI]: 1.8-4.6, p<0.0001) and mortality (HR: 3.8, 95%CI: 1.4-10, p<0.01). When compared with Fontan patients with normal UA with low BNP, those with HUA and high BNP (≥18.4 pg/ml) had a high HR of 7.0 (CI:3.5-14.1, p<0.0001) and 11.7 (CI:2.9-78.1, p<0.001) for USH and all-cause mortality, respectively.
Conclusions: UA is a simple and clinically useful marker for stratifying heart failure severity of Fontan patients and HUA predicts the morbidity and mortality.
Article Information
vol. 130 no. Suppl 2 A11572
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Hideo Ohuchi;
- Yosuke Hayama;
- Jun Negishi;
- Kanae Noritake;
- Osamu Sasaki;
- Nobuyuki Tsujii;
- Toru Iwasa;
- Heima Sakaguchi;
- Aya Miyazaki;
- Osamu Yamada
- Pediatric Cardiology, National Cerebral and Cardiovascular Cntr, Osaka, Japan
Abstract 11621: Fibrillin-1 Gene Mutations in Left Ventricular Non-Compaction Cardiomyopathy
John J Parent, Jeffrey A Towbin, John L Jefferies
Circulation. 2014;130:A11621
Abstract
Introduction: Left ventricular non-compaction cardiomyopathy (LVNC) is a rare and unique cardiomyopathy. Its presentation can range from a benign phenotype to overt heart failure and sudden cardiac death. The genetics of LVNC are not completely understood and current genetic testing has a yield of about 30% in identifying a causative gene mutation. We present a series of patients with LVNC and fibrillin-1 (FBN1) gene mutations.
Hypothesis: We hypothesize that FBN1 gene mutations can lead to LVNC by way of its role in the myocardial extracellular matrix during cardiac development.
Methods: A retrospective review of all patients with LVNC at our institution was performed for purposes of another investigation. The process unexpectedly identified patients with LVNC and FBN1 gene mutations, as well as LVNC and Marfan syndrome.
Results: Approximately 150 patients are followed in our clinic with LVNC. We screened this population and found 51 patients on medical therapy for reduced function. We retrospectively reviewed gene testing in these 51 patients, when available, and identified 5 patients (10%) with an FBN1 gene mutation. All 5 patients had a dilated LVNC phenotype and previous or current evidence of left ventricular dysfunction. Syndrome breakdown as follows: 3 with Marfan, 1 with Shprintzen-Goldberg, and 1 with no identifiable syndrome. Dilated cardiomyopathy/LVNC gene testing was performed in 3 patients; 2 had disease causing myosin heavy chain 7 gene defects and 1 had no defects.
Conclusions: The role of FBN1 in the human myocardium is not completely understood but it is expressed in the developing fetal heart and is a component of the myocardial extracellular matrix. Although causation has not been proven by our report, it certainly raises interest in a mechanistic relationship between LVNC and FBN1 given the increased prevalence of Marfan syndrome and probable increased prevalence of FBN1 gene mutations in this cohort of LVNC patients in light of FBN1.
Article Information
vol. 130 no. Suppl 2 A11621
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- John J Parent;
- Jeffrey A Towbin;
- John L Jefferies
- Pediatric Cardiology, Cincinnati Children’s Hosp Med Cntr, Cincinnati, OH
Abstract 20424: Extracorporeal Membrane Oxygenation (ECMO) Support in Single Ventricle Patients After the Glenn Procedure: Review of the Extracorporeal Life Support Organization (ELSO) Registry
Scott I Aydin, Melissa Duffy, Daniel Rodriguez, Ravi R Thiagarajan, Samuel Weinstein
Circulation. 2014;130:A20424
Abstract
Introduction: ECMO support has been used for both acquired and complex congenital cardiac disease. Despite additional technical challenges, there has been an increase in the use of ECMO for complex congenital cardiac patients with single ventricle physiology. While few reports have been published on the use of ECMO following the Norwood or Fontan operations, only case series on its use following the Glenn shunt exist.
Methods: Data was collected from the ELSO Registry for patients with a Glenn shunt supported with ECMO from 1990-2012. Demographics, survival, and data pertaining to ECMO support were obtained. Descriptive statistics and multivariate analyses utilizing Mann-Whitney U, chi-square, or Fisher’s exact tests were performed to identify associations with mortality.
Results: Two hundred twenty nine patients with bidirectional or classic Glenn shunts had ECMO performed at a median age of 5.9 months [4.4, 8.8]. Hospital survival was 33%. Primary indications were cardiac for 162 patients (71%), respiratory for 27 (12%) and ECPR for 40 (17%). Survival was 33% for cardiac, 37% for respiratory and 30% for ECPR indications. Univariate analysis demonstrated pre-ECMO variables of lower weight (5.8 kg versus 6.2 kg; p = 0.03), elevated PaCO2 (55 mmHg versus 44 mmHg; p < 0.005), lower PaO2 (32 mmHg versus 37 mmHg; p = 0.05), use of bicarbonate (p = 0.02), and longer duration of intubation (34 hours versus 15 hours; p = 0.003) as significant associations with mortality. ECMO variables significantly associated with mortality were elevated oxygen concentrations (0.4 versus 0.35; p = 0.03), longer duration of ECMO support (151 hours versus 91 hours; p < 0.001), circuit complications (p = 0.005), neurologic complications (p = 0.05), and renal injury (p < 0.001). Multivariate analysis demonstrated only elevated pre-ECMO PaCO2 (OR 1.022, 95% CI 1.01-1.04; p = 0.008) and renal injury (OR 2.51, 95% CI 1.15-5.53; p < 0.001) as significant associations with mortality.
Conclusion: Despite the challenges presented by complex anatomy, patients following the Glenn shunt for single ventricular physiology can be supported successfully, though overall survival is less than the general population. Hypercarbia prior to ECMO and renal injury were associated with increased mortality.
Article Information
vol. 130 no. Suppl 2 A20424
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Cardiology and Critical Care, The Children’s Hosp at Montefiore, Bronx, NY
- 2Cardiothoracic Surgery, Montefiore Med Cntr, Bronx, NY
- 3Perfusion, Montefiore Med Cntr, Bronx, NY
- 4Cardiology, Boston Children’s Hosp, Boston, MA
Abstract 18813: Outcomes of Patients with Congenital Heart Disease following Heart Transplantation: the Impact of Disease Type, Prior Thoracic Surgeries and End-Organ Dysfunction
Matthew J Lewis, Jonathan Ginns, P. C Schulze, Matt Lippel, Paul Chai, Emile Bacha, Donna Mancini, Marlon Rosenbaum, Mary Jane Farr
Circulation. 2014;130:A18813
Abstract
Introduction: Adults with congenital heart disease (ACHD) are at increased risk for early adverse outcomes following heart transplantation (Htx). Despite the need for improved risk stratification, small cohorts have constrained identification of patient-specific factors associated with poor prognosis. We hypothesized that type of CHD, number of sternotomies and prior end-organ dysfunction would be associated with an increased risk for mortality post-HTx.
Methods: We performed a retrospective, observational cohort study of all patients with ACHD who underwent HTx at our institution from 1/1997 to 1/2014. The primary endpoint was death. Exposures of interest included type of CHD, number of sternotomies and hepatic dysfunction secondary to passive congestion, measured as high Models for End-Stage Liver Disease Excluding INR (MELD-XI) score.
Results: 48 ACHD patients were followed (mean age at HTx: 36±1.7 years). Diagnoses included: Tetralogy of Fallot (TOF)/pulmonary atresia/double outlet right ventricle in 15 (31%), D-transposition of the great arteries in 10 (21 %), tricuspid atresia/double inlet left ventricle in 9 (19%), VSD/ASD in 4 (8%), heterotaxy in 3 (6%), congenitally corrected transposition of the great arteries in 2 (4%), and 5 (10%) with other diagnoses. Pre-transplant cardiac surgeries included: Fontan operation in 12 (25%), Mustard/Senning repair in 9 (19%), TOF repair in 5 (11%), Rastelli repair in 5 (11%), VSD/ASD closures in 3 (6%), other another congenital heart surgery in 8 (17%) and no prior cardiac surgery in 5 (11%).
Over a median follow-up time of 3.9 years, 14 patients died (29%) and 10 (71%) deaths occurred within 30 days of HTx. Survival at 1 and 5 years was 77%. Deaths within 30 days of surgery were secondary to hemorrhage in 4 (40%), graft failure in 3 (30%) and multi-system organ failure in 3 (30%). By multivariate analysis, ≥3 sternotomies (HR 8.5, p=0.02), MELD-XI score >18 (HR 6.2, p=0.009) and heterotaxy (HR 9.8, p=0.01), were significant predictors of mortality.
Conclusion: In our cohort of patients with CHD undergoing Htx, ≥3 sternotomies, MELD-XI score >18 and heterotaxy syndrome were significantly associated with death. These findings may be important in patient selection and timing of Htx in this population.
Article Information
vol. 130 no. Suppl 2 A18813
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Matthew J Lewis1;
- Jonathan Ginns1;
- P. C Schulze1;
- Matt Lippel1;
- Paul Chai2;
- Emile Bacha2;
- Donna Mancini1;
- Marlon Rosenbaum1;
- Mary Jane Farr1
- 1Cardiology, Columbia Univ/New York Presbyterian Hosp, New York, NY
- 2Surgery, Columbia Univ/New York Presbyterian Hosp, New York, NY
Abstract 17968: Increased Risk of Post-Heart Transplant Mortality in Small Infants
Heang M Lim, Justin Godown, Sunkyung Yu, Janet E Donohue, Joshua M Friedland-Little, Robert J Gajarski, Kurt R Schumacher
Circulation. 2014;130:A17968
Abstract
Background: Infants awaiting heart transplant (HTx) continue to have high waitlist and early post-HTx mortality. The impact of small patient size on wait-list and post-HTx outcomes in infants has not been well studied. The objective of this study was to assess the effect of small size at the time of listing on waitlist outcomes, post-HTx mortality, and post-HTx complications.
Methods: Infants weighing < 5kg listed for HTx from 1994-2013 were identified from the Organ Procurement and Transplantation Network database. The cohort was divided into 2 groups: < 3kg and 3-5kg. Waitlist duration was compared using Wilcoxon rank sum test. Waitlist mortality was examined using competing risk analysis, with delisting as a competing risk. Cox proportional hazard model assessed the impact of size at listing on post-HTx 1-year mortality.
Results: Of 1,966 infants included in the analysis, 442 (22.5 %) weighed < 3 kg, and 1,524 (77.5 %) weighed 3-5 kg at listing. Between the two groups, there was no significant difference in waitlist duration (p=0.24) or waitlist mortality (Adjusted hazard ratio (AHR) 1.13, 95% confidence interval (CI) 0.90, 1.40; p=0.29). Weight < 3kg was an independent risk factor for 1-year post-HTx mortality (AHR 1.53, 95% CI 1.14, 2.05; p=0.01), despite similar donor-to-recipient weight ratios. Post-HTx complications including infection, dialysis, stroke, and the need for re-operation were found to be similar despite patient size.
Conclusions: Weight < 3 kg at listing did not affect waitlist duration or waitlist mortality but was an independent risk factor for 1-year post-HTx mortality. These findings suggest that small infants can be effectively supported to HTx, but small size must be considered when assessing an infant’s overall risk for post-HTx mortality.
Article Information
vol. 130 no. Suppl 2 A17968
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Heang M Lim;
- Justin Godown;
- Sunkyung Yu;
- Janet E Donohue;
- Joshua M Friedland-Little;
- Robert J Gajarski;
- Kurt R Schumacher
- Michigan Congenital Heart Cntr, C.S. Mott Children’s Hosp, Univ of Michigan, Ann Arbor, MI
Abstract 18139: Myocardial Fibrosis Quantified by Cardiovascular Magnetic Resonance is Associated with Histology and Graft Function After Pediatric Heart Transplantation
Brian Feingold, Cláudia M Salgado, Miguel Reyes-Múgica, Stacey Drant, Susan A Miller, Peter Kellman, Erik B Schelbert, Timothy C Wong
Circulation. 2014;130:A18139
Abstract
Background: Late survival after pediatric heart transplantation (HTx) remains poor. Many late deaths are due to “graft failure,” typically in the presence of vasculopathy and diffuse myocardial fibrosis (DMF) – a process associated with ventricular remodeling and heart failure (HF). Cardiovascular magnetic resonance (CMR)-derived extracellular volume (ECV) is a validated measure of DMF in the absence of edema or infiltrative disease, and predicts outcomes of HF and mortality in adults. We hypothesize that ECV is a meaningful biomarker of graft dysfunction following pediatric HTx.
Objective: To test the association of ECV with histologic myocardial fibrosis after pediatric HTx. We also explored associations of ECV with hemodynamic, echocardiographic, and serum measures of graft function.
Methods: We prospectively enrolled consecutive HTx recipients who were ≥13 years old and ≥9 months post HTx for ECV quantification at the time of surveillance endomyocardial biopsy (EMB). Fibrosis was quantified on EMB by automated image analysis after picrosirius staining and digital scanning. CMR measures of blood and myocardial T1 from basal and mid short axis slices, along with contemporaneous hematocrit, quantified ECV.
Results: Nineteen pts (12 male) underwent CMR at a mean age of 18.4 ± 2.8 yrs (range 14.9 – 24.4 yrs) and a mean time after HTx of 10.4 ± 6.6 yrs (1.0 – 20.7 yrs). Four pts were excluded from analysis due to acute rejection (ISHLT grade ≥2R) on concurrent EMB (n=2) or poor quality imaging (n=2). Mean ECV was 27.1 ± 3.8 (20.9 – 32.1). Late gadolinium enhancement was observed in 1 pt. ECV showed moderate correlations with histologic myocardial fibrosis (r=0.61; p=0.02) and serum b-type natriuretic peptide (r=0.66; p=0.008). There was a trend to correlation with pulmonary capillary wedge pressure (r=0.51; p=0.06). We found no associations of ECV with systolic or diastolic function, time after HTx, or graft age.
Conclusions: We demonstrate a novel association of ECV with histologic myocardial fibrosis and serum and hemodynamic markers of HF after pediatric HTx. Given prior observations of myocardial fibrosis in chronic graft failure, these findings suggest that ECV may be a relevant, noninvasive marker of graft dysfunction and a potential therapeutic target.
Article Information
vol. 130 no. Suppl 2 A18139
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Brian Feingold1;
- Cláudia M Salgado2;
- Miguel Reyes-Múgica2;
- Stacey Drant1;
- Susan A Miller1;
- Peter Kellman3;
- Erik B Schelbert4;
- Timothy C Wong4
- 1Pediatric Cardiology, Children’s Hosp of Pittsburgh of UPMC, Pittsburgh, PA
- 2Pathology, Children’s Hosp of Pittsburgh of UPMC, Pittsburgh, PA
- 3Laboratory of Cardiac Energertics, National Heart Lung and Blood Institute, NIH, Bethesda, MD
- 4Medicine, Univ of Pittsburgh Sch of Medicine, Pittsburgh, PA
Abstract 17400: Modern Outcomes of Mechanical Circulatory Support as a Bridge to Pediatric Heart Transplantation
Brody Wehman, Kristen Stafford, Gregory Bittle, Zachary N Kon, Amelia C Watkins, Nicholas Pietris, Osama T Siddiqui, Keshava Rajagopal, Si Pham, Sunjay Kaushal, Bartley P Griffith
Circulation. 2014;130:A17400
Abstract
Introduction: Pediatric patients awaiting heart transplantation frequently require bridge to transplantation (BTT) with mechanical circulatory support (MCS). Long-term survival outcomes and predictors of mortality have yet to be described in the modern era using a large-scale analysis.
Hypothesis: We hypothesized that compared to DTXP, children BTT with ECMO would have inferior post-transplant survival.
Methods: The United Network for Organ Sharing database was reviewed to identify pediatric heart transplantation recipients from 2005 through 2012. Patients were stratified into 3 groups: ECMO, VAD or DTXP. The primary outcome was post-transplant survival.
Results: 2,777 pediatric heart transplantation recipients were identified. There were 617 patients BTT with MCS (22.2%), of whom there were 428 VAD BTT (69.4%) and 189 ECMO BTT (30.6%). An increase in VAD use was observed over the study period (P<0.0001). Compared to DTXP, ECMO BTT patients had a lower median age (<1 vs. 5 years, P<0.0001) and were significantly smaller (8 vs.14 kg, p<0.001), while VAD BTT patients were older (8 vs. 5 years, P=0.0002) and larger (24 vs. 14 kg, P<0.001). Actuarial survival was greater in DTXP compared to ECMO BTT, but similar to VAD BTT at 30 days, 1, 3 and 5 years (P<0.0001) (Figure 1). After censoring the first 4 months post-transplant, survival in the ECMO BTT and VAD BTT groups were equivalent to DTXP. In multivariable analysis, ECMO BTT was an independent predictor of post-transplant mortality (HR 2.77, CI 2.12-3.61, P<0.0001), while VAD BTT was not (HR 1.20, CI 0.91-1.58, P=0.15).
Conclusions: Pediatric patients with DTXP or VAD BTT have equivalent post-transplant survival. However, those requiring ECMO BTT have inferior post-transplant survival compared to those receiving DTXP.
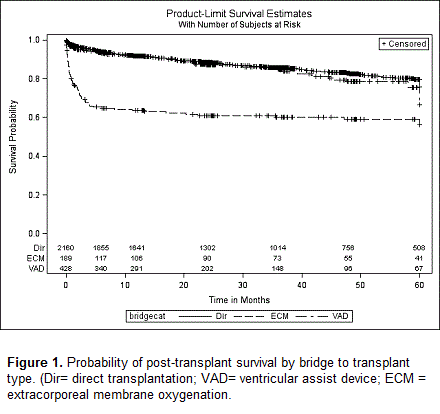
Article Information
vol. 130 no. Suppl 2 A17400
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Brody Wehman1;
- Kristen Stafford2;
- Gregory Bittle1;
- Zachary N Kon1;
- Amelia C Watkins1;
- Nicholas Pietris3;
- Osama T Siddiqui1;
- Keshava Rajagopal1;
- Si Pham1;
- Sunjay Kaushal1;
- Bartley P Griffith1
- 1Surgery, Univ of Maryland Med Cntr, Baltimore, MD
- 2Epidemiology and Public Health, Univ of Maryland Sch of Medicine, Baltimore, MD
- 3Pediatrics, Univ of Maryland Med Cntr, Baltimore, MD
Abstract 17587: Myocarditis is Not an Independent Risk Factor for Outcomes in Children Listed for Heart Transplantation
Jessica C Garbern, Kimberlee Gauvreau, Elizabeth D Blume, Tajinder P Singh
Circulation. 2014;130:A17587
Abstract
Introduction: While many children with myocarditis recover, some have persistent heart failure and are listed for heart transplantation (HT). Previous reports suggest worse post-transplant survival in children with myocarditis compared to children with dilated cardiomyopathy (DCM) leading some to theorize that viral/inflammatory factors adversely affect graft survival.
Hypothesis: Among children listed for HT, those with myocarditis have higher wait-list and post-transplant mortality compared to children with DCM; however, these differences are accounted for by differences in baseline risk factors.
Methods: We identified all US children < 18 yrs. of age between 07/04 and 12/10 who were listed for HT with a diagnosis of myocarditis or DCM in the Organ Procurement and Transplant Network database. Cox models were used to determine whether myocarditis is independently associated with wait-list mortality (or becoming too sick to transplant) and post-transplant graft loss (death/re-HT).
Results: Overall, 139 children with myocarditis and 1058 with DCM were analyzed. Children with myocarditis were more likely to be listed as status 1A (81% vs. 71%, P=0.02), on ventilator support (48% vs. 25%, P<0.001), mechanical support (44% vs. 18%, P<0.001) and with renal dysfunction (52% vs. 42%, P<0.01). Children with myocarditis were more likely to die on the wait-list or become too sick to transplant (18% vs. 10%, hazard ratio (HR) 2.0, 95% CI 1.3-3.1). However, in analysis adjusted for age, listing status, ventilator support and renal function at listing, myocarditis was not associated with wait-list mortality (HR 1.4, 95% CI 0.9-2.2, P=0.16). Overall, 86 children with myocarditis and 824 with DCM received HT. Children with myocarditis were at higher risk of graft loss compared to children with DCM (HR 1.8, 95% CI 1.0-3.0). However, in analysis adjusted for age, race, mechanical support and renal function at HT, myocarditis was not associated with risk of graft loss (HR 1.4, 95% CI 0.8-2.4, P=0.27).
Conclusions: Among children listed for HT, those with myocarditis are at higher risk of wait-list and post-transplant mortality compared to children with DCM. However, these differences in outcomes may be explained by their differences in severity of illness at listing/HT.
Article Information
vol. 130 no. Suppl 2 A17587
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Jessica C Garbern;
- Kimberlee Gauvreau;
- Elizabeth D Blume;
- Tajinder P Singh
- Cardiology, Boston Children’s Hosp, Boston, MA
Abstract 16935: Hyponatremia in Children Hospitalized with Decompensated Heart Failure: Prevalence, Severity and Association with Clinical Outcome
Jack Price, Kevin Chiou, Jonathan Hanna, Jason Goldberg, Timothy Humlicek, Joseph Hagan, Antonio Cabrera, Aamir Jeewa, Susan Denfield, William Dreyer, Ayse Arikan
Circulation. 2014;130:A16935
Abstract
Introduction: Hyponatremia occurs commonly in adults with heart failure (HF) but its prevalence, severity and clinical impact are not known in children.
Hypothesis: Hyponatremia is associated with adverse clinical outcomes in children hospitalized with HF.
Methods: Consecutive patients (pts) from a single institution who required hospitalization for treatment of acute decompensated heart failure were studied. Inclusion criteria: pts age 3 months to 21 years hospitalized for HF attributable to ventricular dysfunction. Exclusion criteria: acute graft rejection, disease of the central nervous system, HF attributable to left-to-right intracardiac shunts, cyanotic heart disease, treatment with vasopressin during hospitalization. Admission serum sodium (Na) concentration and lowest Na before the composite end-point or discharge were examined. The composite end-point was death, transplant or use of mechanical circulatory support.
Results: One hundred forty-one patients met study criteria. Etiologies of HF included: dilated cardiomyopathy (n=89, 63%), acute myocarditis (n=27, 19%), ischemic cardiomyopathy (n=17, 12%), restrictive cardiomyopathy (n=5, 4%), hypertrophic cardiomyopathy (n=5, 4%). The cohort included 48 (34%) pts with pre-existing HF. Mean serum Na at admission was 137 ±4 mmol/L. Hyponatremia (serum Na <135 mmol/L) was present in 45 (32%) pts at admission. Hyponatremia subsequently developed in 72 (51%) pts during their hospitalization and dropped below 130 mmol/L in 50 (35%) pts. The composite end-point occurred in 58 (41%) pts. Those pts who were hyponatremic at admission were more likely to reach the composite end-point than pts who had normal or high Na concentrations at admission (26/45 pts [58%] vs 32/96 [33%], p=0.006). Among pts who met the composite end-point, the mean lowest serum Na concentration was 128 (±4) mmol/L. Hyponatremia at admission was independently associated with death, transplant or the use of mechanical circulatory support during hospitalization (OR 3.1, p=0.003).
Conclusions: Hyponatremia occurs commonly in children hospitalized with acute decompensated HF and is associated with in-hospital mortality, transplant and the need for mechanical circulatory support.
Article Information
vol. 130 no. Suppl 2 A16935
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Jack Price;
- Kevin Chiou;
- Jonathan Hanna;
- Jason Goldberg;
- Timothy Humlicek;
- Joseph Hagan;
- Antonio Cabrera;
- Aamir Jeewa;
- Susan Denfield;
- William Dreyer;
- Ayse Arikan
- Pediatric Cardiology, Texas Children’s Hosp, Houston, TX
Abstract 16939: Pediatric Quality of Life While Supported With a Ventricular Assist Device
Jacob R Miller, Umar S Boston, Deirdre J Epstein, Matthew C Henn, Christopher P Lawrance, Jacob Kallenbach, Kathleen E Simpson, Charles E Canter, Pirooz Eghtesady
Circulation. 2014;130:A16939
Abstract
Introduction: The Ventricular Assist Device (VAD) has emerged as an important treatment option for bridging pediatric patients with heart failure to transplant. VADs have been shown to improve survival; however, quality of life (QoL) while supported with a pediatric VAD is unknown.
Methods: In this prospective study, pediatric patients, and their parents, who underwent VAD placement completed a generic PedsQL 4.0 biweekly until heart transplant or death. Data are reported at last follow-up with a VAD and compared to three previously reported groups: age-matched healthy controls, outpatients with severe heart disease and children after heart transplant.
Results: From December 2007 to August 2013, 13 patients received VAD support for at least two weeks and completed a PedsQL. The mean age at implant was 10.0 ± 4.2 years with a median duration of support of 1.6 months (Table 1). Pre-implant INTERMACS profile was 1 for 3/13 (23%) and 2 for 10/13 (77%). Patients self-reported significantly (p<0.05) reduced total and physical QoL scores when compared to each of the three comparison groups. Self-reported psychosocial QoL scores were significantly lower than healthy controls only. Parent proxy-reported scores were significantly lower than all three comparison groups for all three categories (Figure 1).
Conclusion: A large deficit exists in the QoL of pediatric patients supported by a VAD compared to outpatient management of severe heart disease or post-heart transplant patients; however, VAD patients do represent a sicker group. Improvements in QoL must be made, as time spent with a VAD is expected to increase. Though a small sample, these data can serve as a baseline for future comparison.
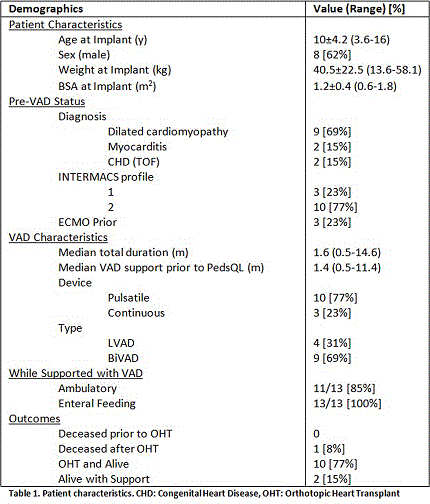
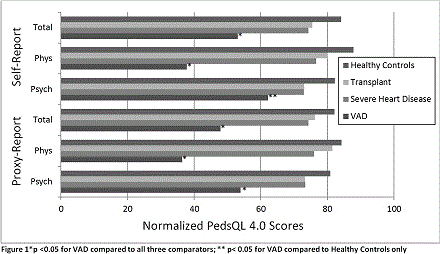
Article Information
vol. 130 no. Suppl 2 A16939
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Jacob R Miller1;
- Umar S Boston2;
- Deirdre J Epstein2;
- Matthew C Henn1;
- Christopher P Lawrance1;
- Jacob Kallenbach2;
- Kathleen E Simpson3;
- Charles E Canter3;
- Pirooz Eghtesady2
- 1Surgery, Washington Univ in St. Louis, St. Louis, MO
- 2Pediatric Cardiothoracic Surgery, St. Louis Children’s Hosp, St. Louis, MO
- 3Pediatric Cardiology, St. Louis Children’s Hosp, St. Louis, MO
Abstract 11079: Medical Therapy Leads to Positive Remodeling in Left Ventricular Non-Compaction Cardiomyopathy
John J Parent, Jeffrey A Towbin, John L Jefferies
Circulation. 2014;130:A11079
Abstract
.
Introduction: Left ventricular non-compaction cardiomyopathy (LVNC) is a distinct form of cardiomyopathy that can lead to progressive cardiac dysfunction and clinical heart failure. LVNC with left ventricular dilation or dysfunction is associated with a greater risk for mortality.
Hypothesis: We hypothesized that initiation of heart failure medications in patients with LVNC and ventricular dysfunction, with or without dilation, would improve systolic function and reduce ventricular dilation.
Methods: The study was a retrospective chart review. Inclusion criteria were as follows: presence of LVNC, reduced systolic function [Left ventricular ejection fraction (EF) < 55% or shortening fraction (SF) < 30%], therapy with at least one medication (beta blocker, ACE inhibitor, ARB), imaging performed both pre- and post-initiation of therapy.
Results: Fifty one patients met inclusion criteria. Forty eight had complete echocardiographic data and 8 had complete cardiac MRI data. Mean age at initiation of medication was 11.5 ± 11.8 years. Follow-up, defined as time from initiation of medication to most recent echocardiogram, was 2.4 ± 2.3 years. Three patients (6%) were solely on a beta blocker, 15 (29%) were on ACE/ARB monotherapy, and 33 (65%) were on dual therapy with a beta blocker and an ACE/ARB. After initiation of medical therapy 38/44 (86%) had improvement in EF by ≤ 5%, 27/40 (68%) had improvement in their SF by ≤ 5%, 6/44 (14%) had no change in EF, and 11/40 (28%) had no change in SF. No patient (0/44, 0%) had a decline in EF by ≤5%, but 2/40 (5%) had a drop in SF by ≤ 5%.
A two-sided paired t-test was performed comparing EF, SF, and left ventricular end-diastolic dimension (LVEDD) in the cohort before and after therapeutic intervention demonstrating a 16 ± 12% improvement in EF (p < 0.0001), an 8 ± 9% improvement in SF (p < 0.0001), and a 0.83 ±1.93 (p<0.05) decrease in LVEDD z-score.
Conclusions: Early diagnosis and medical treatment of LVNC with reduced systolic function leads to favorable left ventricular remodeling evident by an improvement in left ventricular systolic function and reduction of LVEDD.
Article Information
vol. 130 no. Suppl 2 A11079
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- John J Parent;
- Jeffrey A Towbin;
- John L Jefferies
- Pediatric Cardiology, Cincinnati Children’s Hosp Med Cntr, Cincinnati, OH
Abstract 20667: Cardiomyocyte miR-29 Promotes Cardiac Remodelling
Yassine Sassi, Petros Avramopoulos, Deepak Ramanujam, Bart De Strooper, Stefan Engelhardt
Circulation. 2014;130:A20667
Abstract
Cardiac hypertrophy is the response of the heart to several stimuli such as myocardial infarction and volume or pressure overload. MicroRNAs have been shown to play an important role in cardiac disease. miR-29 is predicted to target a number of extracellular matrix genes including collagens and matrix metallopeptidases (MMP). Whereas previous studies have focused on the role of miR-29 in cardiac fibroblasts and in the healthy heart, the role of miR-29 in cardiomyocytes and during cardiac disease remained unclear. In the present study we investigated the role of miR-29 in the myocardium by chemical and genetic manipulation of miR-29 in vitro and in vivo.
We found that miR-29 was preferentially expressed in cardiomyocytes rather than in cardiac fibroblasts in adult rodents and that their expression was transiently increased during the early phase of cardiac hypertrophy induced by transverse aortic constriction (TAC) in mice. Furthermore miR-29 promoted hypertrophic growth of primary cardiomyocytes in vitro (CM mean cell area (μm2): miR-control 136±5; miR-29a: 290±30) whereas antagonization of miR-29 with locked nucleic acid-modified antisense molecules (LNA-29) efficiently impeded phenylephrine-induced hypertrophy in vitro. Genetic deletion of miR-29 in gene targeted mice prevented cardiac hypertrophy and fibrosis induced by TAC. To overexpress miR-29 specifically in cardiomyocytes, an AAV9 construct carrying miR-29a (AAV-miR-29a) was injected intravenously into mice that were subjected to TAC or Sham surgery. AAV-miR-29a significantly increased TAC-induced cardiac hypertrophy (heart weight-to-tibia length ratio), which was paralleled by an increase of the average cardiomyocyte cross-sectional area. Conversely, LNA-29 protected mice from cardiac remodeling induced by TAC (HW/TL ratio: Sham: 5.27±0.3; TAC control: 9.4±0.1; TAC LNA-29: 6.5.±0.4). Finally, a series of bioinformatic and biochemical analyses revealed that miR-29 directly targets the GSK3β/NFAT pathway in cardiomyocytes and thereby promotes cardiomyocyte hypertrophy.
Together, our results indicate the members of the miR-29 family as important regulators of cardiomyocyte hypertrophy and as potential therapeutic targets in myocardial disease.
Article Information
vol. 130 no. Suppl 2 A20667
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Technische Universität München, Institut für Pharmakologie und Toxikologie, Munich, Germany
- 2Cntr for Human Genetics, VIB Cntr for the Biology of Disease, Leuven, Belgium
Abstract 20451: Hedgehog Signaling and Cardiomyocyte Proliferation
Bhairab N Singh, Stefan M Kren, Wuming Gong, Cyprian Weaver, Kathy Bowlin, Elizabeth Braunlin, Mary G Garry, Naoko Koyano-Nakagawa, Daniel J Garry
Circulation. 2014;130:A20451
Abstract
Background: In contrast to adult mammalian heart, lower vertebrates such as newt exhibit a dramatic capacity for regeneration in response to injury. Understanding the underlying mechanisms and signaling pathways in newt could form the basis for regenerative therapies in mammals. In the present study, we explored the role of hedgehog (HH) signaling during cardiac regeneration.
Methods and Results: To investigate cardiac regeneration (CR) in vivo, we performed ventricular resection studies in adult newt heart. Whole mount and histochemical studies revealed CR within 30 d of resection injury. Functional assessment by echocardiographic analysis showed improved ejection fraction from 22 ± 5% at 4 d to 42 ± 3% at 30 d of regeneration relative to control (60 ± 3%). Gene clustering and qRT-PCR analysis at 7d post injury indicated enrichment of hedgehog (HH) signaling factors including Shh and ptch1 by 3- and 2-fold respectively, suggesting a critical requirement of HH signaling at early stages of CR. We determined HH signaling is essential as pharmacological inhibition of the HH pathway resulted in complete ablation of CR. Using EdU-labeling and immunohistochemical analyses, we showed that HH signaling regulates proliferation of both epicardial and myocardial cells, as inhibition of the HH pathway reduced EdU-positive nuclei from 15 ± 2% to 5 ± 3%. In contrast, qRT-PCR analysis from murine hearts postnatal day (P) 2 to P14, showed reduced levels of HH signaling factors and cell-cycle associated mRNAs by 3.5-fold with concomitant increase of p21 by 2-fold. This implies HH signals are required for the proliferative process. Consistent with the newt studies, activation of HH in murine neonatal ventricular cardiomyocytes (NVCM) promoted cardiomyocyte proliferation, increasing positive nuclei from 10 ± 2% to 25 ± 3%, while inhibition of HH signaling reduced positivity to 6 ± 3%. qRT-PCR analysis established that activation of HH signaling in NVCM resulted in up regulation of transcripts associated with cardiomyocyte proliferation.
Conclusion: These data indicated that HH signaling pathways modulate cardiomyocyte proliferation providing potential therapeutic targets for achieving mammalian cardiac regeneration.
Article Information
vol. 130 no. Suppl 2 A20451
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Bhairab N Singh;
- Stefan M Kren;
- Wuming Gong;
- Cyprian Weaver;
- Kathy Bowlin;
- Elizabeth Braunlin;
- Mary G Garry;
- Naoko Koyano-Nakagawa;
- Daniel J Garry
- Medicine, Univ Of Minnesota, Minneapolis, MN
Abstract 20471: Thrombospondin-4 is a Key Mediator in Cardiac Aging
Dong I Lee, San S Yu, Djahida Bedja, David A Kass, Oscar H Cingolani
Circulation. 2014;130:A20471
Abstract
Thrombospondin-4 (TSP4) is a matricellular protein that is essential for the heart to adapt to stress. We previously demonstrated that mice lacking TSP4 develop heart failure (HF) when exposed to high blood pressure, the latter phenomenon being secondary to deficient extracellular matrix (ECM)-myocyte crosstalk. In the present study we tested the hypothesis that TSP4 is also a key mediator in cardiac aging. TSP4-/- mice and controls (WT) were followed for almost 3 years. TSP4 expression increased with aging as well as with exercise. TSP4-/- died at a younger age than WT (Kaplan-Meyer curve; p= 0.001). TSP4-/- developed HF (LVEF = 58 ± 12 vs 38 ± 14 %, for WT and TSP4-/-; p< 0.05), and a phenotype characterized by obesity, sedentarism and cardiac fibrosis. A subgroup of old (24 months of age) TSP4-/- and WT animals (N= 8 each) were exposed to intense exercise (treadmill twice daily for 8 weeks). WT mice improved cardiac contractility and LV mass with exercise as opposed to TSP4-/- animals that failed to do so (echo and cardiac pressure-volume analysis; p < 0.05 for LVEF, PRSW and dP/dt/IP, WT vs. TSP4-/-). Isolated cardiac myocyte studies from these animals showed that TSP4-/- contractility and calcium transients failed to augment with isoproterenol p<0.02 WT vs TSP4-/-). Phosphorylation of AMPK was increased in TSP4 -/- hearts, together with phosphorylation of GSK-3 alpha and beta isoforms, effects that seem to be mediated through an interaction between TSP4 and the CD 44 receptor. We report for the first time that TSP4 is essential for aging. Lack of this protein shortens lifespan in mice and reveals a phenotype of decreased cardiac contractility, increased cardiac interstitial fibrosis, with decreased myocyte calcium transients. These effects are present in the isolated myocyte (ECM-independent) and seem to involve AMPK and GSK-3.
Article Information
vol. 130 no. Suppl 2 A20471
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Dong I Lee;
- San S Yu;
- Djahida Bedja;
- David A Kass;
- Oscar H Cingolani
- Medicine/Cardiology, Johns Hopkins Med Insts, Baltimore, MD
Abstract 20492: Mlf1 Regulates Myocyte Proliferation in Postnatal Hearts
Shalini Muralidhar, Feng Xiao, Suwannee Thet, Hesham Sadek
Circulation. 2014;130:A20492
Abstract
Lower vertebrates, such as newt and zebrafish, retain a robust cardiac regenerative capacity following injury. Although adult mammals lack this cardiac regenerative potential, there is ample interest in understanding how heart regeneration occurs, and to reawaken this process in adult humans. Recently, we showed that mice are capable of regenerating their hearts shortly after birth following injury. This regenerative response is associated with robust proliferation of cardiomyocytes without significant hypertrophy or fibrosis. However, this regenerative capacity is lost by 7 days postnatally, coinciding with cell cycle arrest. In an effort to determine the mechanism of cardiomyocytes cell cycle arrest after the first week of life, we performed a gene array after cardiac injury at multiple post-natal time points. This enabled us to identify a number of transcription factors that are differentially expressed during this postnatal window. We recently reported that one of these transcription factors Meis1 regulates postnatal cell cycle arrest of cardiomyocytes. Furthermore, Myeloid leukemia factor 1 (Mlf1), a bhlh transcription factor that has not been previously studied in the heart has similar dysregulated pattern following injury. Our preliminary data with in-vitro knockdown of Mlf1 in cardiomyocyte resulted in 2-fold increase in cardiomyocyte proliferation. Furthermore, immunohistochemistry results indicated that the endogenous expression and nuclear localization of Mlf1 in the post-natal cardiomyocytes coincides with cell cycle arrest. To explore this pattern, we generated a cardiomyocyte-specific Mlf1 knockout mouse, and showed that loss of Mlf1 results in robust cardiomyocyte proliferation in postnatal hearts (P14). Additionally, we confirmed previous reports that Mlf1 regulates p53 and induces cell cycle arrest by induction of CDK inhibitors like p21 and p57 in these Mlf1 KO mice. This suggests a role of Mlf1 in promoting reactivation of injured myocardium through induction of cardiomyocyte proliferation. These findings will further provide evidences of molecular mechanisms involved in the dormant regenerative capacity in adult mammals that can be a potential target of therapeutic approaches.
Article Information
vol. 130 no. Suppl 2 A20492
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Shalini Muralidhar;
- Feng Xiao;
- Suwannee Thet;
- Hesham Sadek
- Internal medicine Cardiology, UT Southwestern Med Cntr, Dallas, TX
Abstract 20575: An Alpha-1A Adrenergic Receptor Agonist Prevents and Treats Heart Failure
Megan D Montgomery, Trevor Chan, Raj Dash, Philip M Swigart, Bat-Erdene Myagmar, Anthony J Baker, Paul C Simpson
Circulation. 2014;130:A20575
Abstract
INTRODUCTION: The neurohormonal hypothesis that sympathetic activation in heart failure (HF) is toxic to the heart led to class I recommended drugs. However, contrary data suggest that sympathetic blockade in HF has limits, such as the negative effects of alpha-1-adrenergic receptor (a1-AR) blockade in humans in ALLHAT and V-HeFT. Conversely, a1-agonists protect myocytes and hearts of multiple species in multiple models through pleiotropic mechanisms, but this is untested in HF in vivo.
HYPOTHESIS: We hypothesized that a1-AR agonism can treat HF in vivo.
METHODS: We screened drugs in neonatal rat and adult mouse ventricular myocytes (NRVM, AMVM), measured telemetry blood pressure (BP), and did subcutaneous infusion in mice with HF from cardiotoxin (doxorubicin, DOX) and pressure overload (TAC).
RESULTS: In NRVMs, the a1A agonist A61603 (A6) was the most efficacious and potent of 10 a1 agonists in ERK activation (EC50 6±1 nM, Emax 22±7) and protein synthesis (EC50 10±1 nM, Emax 1.7±0.1), and EC50s matched a1A binding affinity. In AMVMs, A6 activated ERK (EC50 39±5 nM) and protected against DOX (EC50 15±8 nM, Emax 35±6), an effect lost in the a1A KO. In isolated hearts, A6 activated ERK target genes. A6 10 ng/kg/d infused sc for 7 days had no effect on BP, but increased cardiac pERK (EC50 0.1±0.1 μg/kg/d). With DOX, A6 10 ng/kg/d started at the same time enhanced survival (A6+DOX 84%, n=21; Veh+DOX 34%, n=49). A6 also preserved function (A6 FS 61±2% vs. Veh 49±3%), and reduced serum CK (571±67 vs. 1204±98), TUNEL staining (0.5±0.1 vs. 1.3±0.2), and Sirius Red staining (25±1 vs. 32±1) (n=3-5). A6 effects on survival and FS after DOX were lost in the a1A KO. With TAC (gradient 109 mmHg), FS fell over 2 weeks to 40±1%. A6 from weeks 2 to 4 increased FS (to 51±2%, n=30), while FS remained low with Veh (39±2%, n=24). A6 after TAC did not change heart or myocytes size, but did increase pERK and bMyHC, and reduced cardiac troponin I (A6 5±2 vs. 11±2), cleaved caspase 3 (77±7% of Veh), collagens I and III (46±5% and 56±8% of Veh), and Sirius Red staining (5±4% vs. 9±8%) (n=3-22).
CONCLUSIONS: The high affinity a1A-AR agonist A61603, at a low dose that does not change BP, prevents cell death and fibrosis and improves function and mortality in two mouse models of HF. a1A-AR agonists might be potential HF therapies.
Article Information
vol. 130 no. Suppl 2 A20575
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Megan D Montgomery;
- Trevor Chan;
- Raj Dash;
- Philip M Swigart;
- Bat-Erdene Myagmar;
- Anthony J Baker;
- Paul C Simpson
- Medicine, Cardiology, UCSF/SFVAMC, San Francisco, CA
Abstract 19693: Modeling Adaptive Growth — Biomimetic Culture of Neonatal Aortic Root Explants
Laura M Piechura, Jacques P Guyette, Sarah E Gilpin, Douglas J Mathisen, Harald C Ott
Circulation. 2014;130:A19693
Abstract
Background: Heart valve disease afflicts individuals of all ages, but current treatments, involving replacement with non-viable prosthetics, are particularly suboptimal for young patients. To design “growing” valve replacements, postnatal growth mechanisms must be elucidated; models of adaptive growth are lacking, thereby hindering these investigations. We hypothesized that physiologic mechanical loading with growth factor stimulation would enable the adaptive growth of aortic root explants in vitro. To this end, we designed a biomimetic perfusion bioreactor that permits the concomitant delivery of these factors.
Methods and Results: We constructed a chamber for culturing neonatal rat hearts under pulsatile flow conditions. Parameters were adjusted to mimic native physiology using ultrasound assessment of cusp motion and invasive measurement of left ventricular and aortic pressures. Culture medium was designed by quantification of key growth factor (IGF-1, TGF-β2, VEGF, EGF, BMP-2 and FGF-2) concentrations within age-matched serum via ELISA. Explants were cultured for five days. Controls were explanted from age- and size-matched pups. Dynamically-cultured samples exhibited similar proliferation rates at early time points when compared to those of valve cusps in vivo as assessed by EdU incorporation (10.5% vs. 14.9%, p=0.35). After five days of dynamic culture, native proliferation rates were achieved only with concomitant growth factor administration (13.3% control; 13.1% dynamic+GF; 3.1% dynamic alone). There was no evidence of proliferation in statically-cultured controls.
Conclusions: We describe a novel biomimetic culture system that permits the modeling of adaptive growth in aortic root explants. Our results demonstrate recapitulation of in vivo cell proliferation rates in dynamically-cultured hearts and suggest a role for mechanical and humoral stimulation in mediating aortic valve growth. This system enables the elucidation and manipulation of the mechanisms driving postnatal heart valve growth, thereby informing the design of the ideal pediatric valve graft with adaptive growth potential.
Article Information
vol. 130 no. Suppl 2 A19693
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Sch of Medicine, Stanford Univ, Stanford, CA
- 2Dept of Thoracic Surgery, Massachusetts General Hosp, Boston, MA
Abstract 19542: Endocardial EphB4 Signaling Regulates Neuregulin-1 Expression and is Critically Required for Myocardial Development
Jiang Liu, Binyun Ma, Hai-Yun Yen, Parkash S Gill, Henry M Sucov, S. Ram Kumar
Circulation. 2014;130:A19542
Abstract
The vertebrate ventricular myocardium is formed by a complex and incompletely understood developmental paradigm that regulates accurate organization of an inner trabecular and outer compact zone layers. EphB4, a member of the largest family of receptor tyrosine kinases, is expressed in the developing endocardium, along with its cognate ligand EphrinB2. Whereas EphB4 and EphrinB2 have well-established roles in vascular maturation, their role in cardiac development is unknown. In this study, we show that disruption of embryonic EphB4 expression in the second heart field (Mef2c-cre) or endocardium (Nfat-1c cre) results in a severely thinned and underdeveloped myocardial layer, ventricular non-compaction, heart failure and embryonic lethality around E15.5. Mutant hearts show reduced activation of PI3K pathway and impaired proliferation of developing cardiomyocytes. Knockdown of EphB4 alters multiple downstream signaling pathways of significance in cardiac development. In particular, EphB4 expression regulates neuregulin-1 levels both in vitro and in vivo, which then activates myocardial ErbB receptor signaling. Our study re-iterates the critical role of endocardial signaling in myocardial development. In addition, we identify EphB4 as a novel upstream modulator of Neuregulin-1, and thereby, a critical regulator of myocardial development – a hitherto unrecognized function of this receptor tyrosine kinase.
Article Information
vol. 130 no. Suppl 2 A19542
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Surgery, Univ of Southern California, Los Angeles, CA
- 2Medicine, Univ of Southern California, Los Angeles, CA
- 3Broad Stem Cell Cntr, Univ of Southern California, Los Angeles, CA
Abstract 17028: Genetically-Programmed Suicide of Adrenergic Cells in Mice Produces Left Ventricular Dysfunction as Revealed by High-Resolution Echocardiography
Aaron P Owji, Candice N Baker, Jeffrey L Jacob, Steven N Ebert
Circulation. 2014;130:A17028
Abstract
The enzyme Phenylethanolamine-N-methyltransferase (Pnmt), which catalyzes the conversion of norepinephrine into epinephrine, serves as a marker for adrenergic cells and is expressed in the developing heart. In the present study, we tested the hypothesis that adrenergic cells play an important role in the development of cardiac function by assessing phenotypes of mice with selective ablation of adrenergic cells compared with littermate controls. Briefly, Pnmt-Cre knock-in mice were crossed with ROSA26-eGFP-DTA mice, which results in selective ablation of Pnmt+ adrenergic cells due to activation of the Diphtheria Toxin Alpha (DTA) “suicide” allele from the ROSA26 locus. Control experiments verified that the model was working correctly since Pnmt-Cre/DTA mice had markedly lower Pnmt mRNA (>97% decrease) and fewer Pnmt+ cells (>50% decrease) in adrenal glands compared to littermate controls. Pnmt-Cre/DTA mice were born in normal Mendelian ratios and appeared outwardly normal and healthy at birth and during the early postnatal period, but began to show cardiac deficits by one month of age. High-resolution echocardiography analysis indicated continuing deterioration of cardiac function with age. Mean cardiac output was decreased at four months (10.81±2.73 mL/min n=14 v 18.42±3.37 mL/min n=18, p-value<0.001, Fig. 1), as was ejection fraction (54.34±7.44% v 66.53±8.51%, p<0.01), and fractional shortening (27.64±4.80% v 36.67±7.22%, p<0.01). Moreover, left ventricular function was hypokinetic in the ablated hearts compared to controls, as shown in the M-mode tracings in Fig. 1. These data demonstrate that selective ablation of adrenergic cells leads to impaired cardiac function that progressively worsens with age over the first few months after birth. In conclusion, this novel cellular suicide model is useful for determining how adrenergic cells contribute to the development of cardiac structure and function.
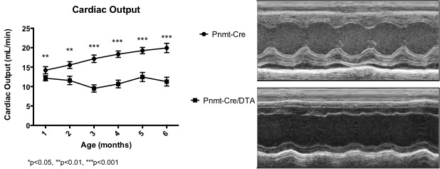
Article Information
vol. 130 no. Suppl 2 A17028
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Aaron P Owji;
- Candice N Baker;
- Jeffrey L Jacob;
- Steven N Ebert
- Burnett Sch of Biomedical Sciences, College of Medicine, Univ of Central Florida, Orlando, FL
Abstract 15907: Disruption of HCN2 Leads to Laterality Defects and Cardiac Arrhythmia
Bin Ye, Greg Taycher, Tim Hacker, Leslie Chan, Nian-Qing Shi
Circulation. 2014;130:A15907
Abstract
Background: Congenital heart defects (CHDs) encompass a large numbers of cardiovascular malformations, and remain the major cause of infant mortality among all types of birth defects. However, molecular mechanisms underlying CHDs remain elusive, largely owning to the complexity of the diseases and lack of animal models that can reproduce the pathophysiological conditions in a laboratory setting. The hyperpolarization-activated, cyclic nucleotide-gated cation channels (HCN) are responsible for generating spontaneous pacemaker activities in cardiac and central nervous systems. These channels are also detected in other cell types such as ventricular myocytes. HCN currents recorded from neonatal cells have an activation threshold of -70 mV while those recorded from adult cells are activated at -110 mV. This difference indicates that HCN activity might be important in early development. However, roles of HCN channels in cardiogenesis and development are not fully understood.
Methods and Results: We created a HCN2 conditional knockout (HCN2KO) model in which the full-length HCN2 was disrupted. Two KO lines were subsequently derived from this model. The first line was weaned by 21 days and they all died by 4-5 wks of age. Maternal ultrasound study revealed that these KO mice developed fetal arrhythmia and had an underdeveloped left side in their hearts. The second line was able to live on under our special diet/care. These mice displayed a slower growth rate (1.1±0.2 g/wk) and lower body weight (15.1±2.0 g) relative to their age-matched WT controls (2.2±0.1 g/wk and 27.5±1.5 g; n=9-10, p<0.05). Echocardiography and tissue staining data suggested that KO hearts had laterality defects compared to their size-matched WT controls. The survived KO mice were able to maintain cardiac function by developing much thicker anterior and posterior walls to sustain blood-pumping (n=6, p<0.05). Electrocardiographic results indicated that the average heart rate recorded from KO mice was ~100 bpm slower than their age-matched WT controls (n=5-6, p<0.05).
Conclusions: These novel findings indicate that HCN2 is indispensable in mouse cardiogenesis and development. Our KO models are therefore innovative platforms for future CHD research.
Article Information
vol. 130 no. Suppl 2 A15907
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Bin Ye;
- Greg Taycher;
- Tim Hacker;
- Leslie Chan;
- Nian-Qing Shi
- Medicine, Univ of Wisconsin, Madison, WI
Abstract 12438: Novel 4-Dimensional Optical Technique to Elucidate Hemodynamic Shear Forces and Initiation of Trabeculation During Cardiac Morphogenesis
Juhyun Lee, Peng Fei, Nelson Jen, Tyler Beebe, Chih-ming Ho, Alison Marsden, Neil Chi, Tzung Hsiai
Circulation. 2014;130:A12438
Abstract
Hemodynamic shear forces are intimately linked with cardiac development. However, the mechano-transduction mechanisms underlying this morphological event remain elusive. Trabeculation develops after cardiac looping, which is visualized in zebrafish embryos starting at 70 hours post-fertilization (hpf). We hypothesized that endocardial wall shear stress (WSS) up-regulates Notch signaling to initiate trabeculation. We developed a novel fast z-axis imaging acquisition system to reconstruct time-dependent 3-dimensional WSS in live zebrafish using single plane illuminating microscopy (SPIM). To assess the link between shear stress and trabeculation, we lowered the WSS by micro-injection of (1) gata1a-morpholino oligonucleotides (MO) at 2-cell stage to reduce erythropoiesis and viscosity by 70%, (2) tnnt2a-MO injection to inhibit cardiac contraction, and (3) use of weak atrium(wea) mutant to inhibit atrial contraction. To determine mechano-signal transduction underlying WSS and trabeculation, we assessed Notch signaling by in situ hybridization of NRG1 and notch1b. At 70 hpf, the control fish showed formation of trabeculation, whereas gata1 MO injected fish revealed a decrease in trabeculation in response to a reduction in shear stress by 90 %. Both wea mutant and tnnt2a MO injected fish exhibited smooth endocardial surfaces without trabecular networking. In situ hybridization of NRG1 and Notch 1b further showed a decrease in fluorescent intensity in the gata1 MO, wea mutant, and tnnt2a MO groups by 2.33±0.87-fold, 4.21±0.25-fold, and 4.96±0.17-fold, respectively (p < 0.05 vs. control fish, n=10). Thus, our findings indicate that hemodynamic shear forces are implicated in the initiation of cardiac trabecualtion via Notch signaling pathway, with a translational implication to understanding congenital noncompacted cardiomyopathy.
Article Information
vol. 130 no. Suppl 2 A12438
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Juhyun Lee1;
- Peng Fei2;
- Nelson Jen1;
- Tyler Beebe1;
- Chih-ming Ho3;
- Alison Marsden4;
- Neil Chi5;
- Tzung Hsiai6
- 1Bioengineering, Univ of California Los Angeles, Los Angeles, CA
- 2Mechanical Engineering, Univ of California Los Angeles, Los Angeles, CA
- 3Mechanical Engineering and Bioengineering, Univ of California Los Angeles, Los Angeles, CA
- 4Mechanical Engineering, Univ of California San Diego, La Jolla, CA
- 5Medicine, Univ of California San Diego, La Jolla, CA
- 6Bioengineering and Medicine, Univ of California Los Angeles, Los Angeles, CA
Abstract 20437: Gene Expression Profiling Identifies Differentially Expressed Genes in Cardiac Allograft Vasculopathy
Monica Colvin-Adams, Nonyelum Harcourt, Ying Zhang, Adam Mitchell, Kenneth Liao, Kenneth Beckman
Circulation. 2014;130:A20437
Abstract
BACKGROUND: Coronary angiography is the current gold standard for screening and diagnosis of cardiac allograft vasculopathy (CAV). Given the invasiveness and marginal sensitivity associated with angiography, minimally invasive sensitive biomarkers are needed to identify CAV early. The goal of our study is to determine differentially expressed genes in CAV that may serve as targets for biomarker development.
Methods: Gene expression profiling and transcriptome analysis using Illumina HiSeq 2000, was performed on 23 cardiac transplant recipients (12 severe CAV ISHLT grade 3 and 11 without CAV) who were matched by age, gender, and duration of transplant. Fastqc, Tophat, Samtools, and CuffDiff were utilized for quality control of raw reads, mapping short reads to reference genomes and testing differential expression between healthy subjects and transplant subject, respectively. Pathway analysis to identify molecular pathways that show expression change in transplant recipients was done using ingenuity pathway analysis (IPA).
Results: Average age was 54.6 years, 69% were men, and the average duration of transplant was 8.1 years. We identified 181 genes termed “big movers”, i.e., genes that show at least two-fold change in expression with raw testing. Of these 181 genes, 149 genes are up-regulated and 32 genes are down-regulated in CAV when compared to nonCAV. MDS plot of expressed genes showed clear separation between CAV and nonCAV samples and further separation in CAV clusters by age. Differentially expressed genes and pathways are shown in table 1.
Conclusion: CAV is associated with differential gene expression in multiple pathways associated with the cell cycle and cell signaling. Elucidation of differentially expressed genes and associated pathways may improve our understanding of CAV and identify targets for early detection of CAV. Longitudinal studies are required to assess the prognostic value of these genes.
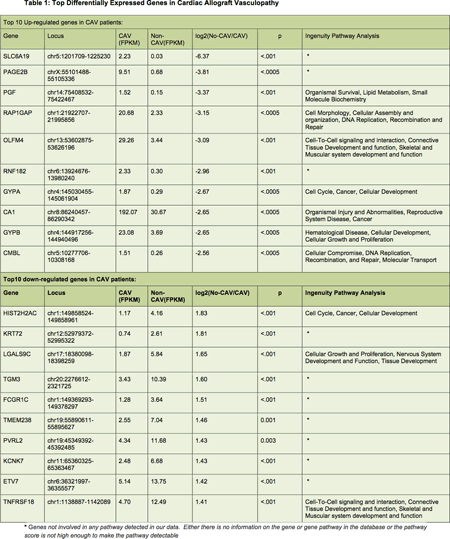
Article Information
vol. 130 no. Suppl 2 A20437
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Monica Colvin-Adams1;
- Nonyelum Harcourt1;
- Ying Zhang2;
- Adam Mitchell3;
- Kenneth Liao4;
- Kenneth Beckman5
- 1Medicine-Cardiology, Univ of Minnesota, Minneapolis, MN
- 2Supercomputer Institute, Univ of Minnesota, Minneapolis, MN
- 3Medicine, Univ of Minnesota, Minneapolis, MN
- 4Surgery, Univ of Minnesota, Minneapolis, MN
- 5Genomics, Univ of Minnesota, Minneapolis, MN
Abstract 20006: RNA Sequencing of Human Hypertrophic Cardiomyopathy Tissue Reveals Diverse Pathways Modulating the Myocardial Tissue Microenvironment
Alejandro de Feria, Simon Maltais, Tarek S Absi, Yan R Su, Thomas P Stricker, Jason R Becker
Circulation. 2014;130:A20006
Abstract
Introduction: Hypertrophic Cardiomyopathy (HCM) is a common inherited cardiac disease and cause of sudden death in adolescents and young adults. Despite significant advances in understanding the genetic underpinnings of HCM, there remains an incomplete understanding of how myofilament mutation carriers and non-carriers ultimately develop myocardial hypertrophy.
Hypothesis: Human hypertrophic cardiomyopathy tissue differentially regulates gene expression involved in cellular growth.
Methods: RNA was extracted from 5 control human septal tissue samples and 8 HCM septal tissue samples. The HCM tissue samples were from both myofilament mutation positive and negative patients. RNA sequencing was performed using Illumina HiSeq 2500 at a depth of 30 million paired-end reads per sample. Sequencing data was aligned to the hg19 genome using TopHat (splice-aware aligner). Cufflinks was used to estimate transcript abundance and differential expression. Pathway enrichment analysis was carried out on significant genes (q<0.05) using DAVID (v6.7). Only pathways with an enrichment score greater than 2 were evaluated.
Results: Cluster and principal component analysis showed highly distinct RNA expression signatures between control and HCM tissues. There were 1500 differentially expressed sequences (q<0.05), which included 1286 annotated genes, 168 novel isoforms, and 46 long noncoding RNA sequences. Gene functional classification revealed 40 differentially regulated gene clusters in HCM tissue compared to controls. The most enriched gene clusters induced in human HCM tissue were associated with secreted peptides involved in promoting cell growth and modulation of extracellular structure (enrichment scores > 10). Highly enriched gene clusters that were suppressed in human HCM tissue were associated with secreted peptides involved in regulating inflammation and immune defense response (enrichment scores > 5).
Conclusion: Our data reveals human HCM tissue differentially regulates a diverse array of genes involved in regulation of the myocardial tissue microenvironment. The strong similarity in gene expression across HCM samples suggests shared pathophysiological mechanisms independent of the underlying genetic etiology.
Article Information
vol. 130 no. Suppl 2 A20006
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- 1Div of Cardiovascular Medicine, Vanderbilt Univ Sch of Medicine, Nashville, TN
- 2Dept of Cardiac Surgery, Vanderbilt Univ Sch of Medicine, Nashville, TN
- 3Dept of Pathology, Microbiology, and Immunology, Vanderbilt Univ Sch of Medicine, Nashville, TN
Abstract 19792: Prediction of Right Ventricular Failure After Cardiac Transplantation : A Recipient Transcriptomic Study
Damien Guijarro, Jean-Pierre Gueffet, Marja Steenman, Jean-Christian Roussel, Jean-Noel Trochu, Guillaume Lamirault
Circulation. 2014;130:A19792
Abstract
Introduction: Right ventricle failure (RVF) is a frequent and severe complication after cardiac transplantation. However, risk stratification for RVF is poorly achieved. Development of transcriptomic biomarkers for outcome prediction in cardio-vascular diseases is promising.
Hypothesis: Our aim was to identify right ventricular gene expression signature associated to RVF and to define a transcriptomic biomarker that could predict post-transplantation RVF.
Methods: Recipient RV myocardial samples of 44 patients transplanted from February 1998 to November 2002 in our center were collected. We retrospectively identified patients with (RVF group) and without (CTL group) post-transplantation RVF.
A 4035-gene expression profile was obtained for all patients. Differentially expressed genes between RVF and CTL groups were identified and a molecular RVF predictor was used to determine for each patient a RVF prediction score (RVFs).
Results: 9 (20%) and 18 (41%) patients were classified in RVF and CTL groups respectively. As compared to CTL group, RVF patients showed higher pre-operative bilirubin level and higher post-operative death rate.
Molecular RVF predictor included 75 differentially expressed genes. Using this predictor, risk for post-transplantation RVF was 2.8-fold greater if RVFs was >0.5 (CI 95%: 1.243-6.305). Sensitivity and specificity of RVFs were 0.778 and 0.889, respectively. Using receiver operating characteristic analysis, RVFs area under curve (AUC) was significantly greater than AUC of commonly used RVF predictors (pulmonary vascular resistance, trans-pulmonary gradient).
Conclusions: Gene expression profiling of recipient right ventricle could be used to predict post-transplantation RVF. Transcriptomic biomarkers should be further investigated as a new tool for selection of cardiac transplant candidates.
Article Information
vol. 130 no. Suppl 2 A19792
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Damien Guijarro1;
- Jean-Pierre Gueffet1;
- Marja Steenman2;
- Jean-Christian Roussel3;
- Jean-Noel Trochu1;
- Guillaume Lamirault1
- 1Cardiology, CHU de Nantes – Institut du Thorax, Nantes, France
- 2Cardiology, Institut du Thorax – Inserm 1087, Nantes, France
- 3Thoracic and Cardiac Surgery, CHU de Nantes – Institut du Thorax, Nantes, France
Abstract 18845: Left Ventricular Assist Device Normalizes Multiple Pathogenic Pathways in Both the Left and Right Ventricle in Patients With Dilated Cardiomyopathy
Nirmal Parajuli, Ratnadeep Basu, Konrad S Famulski, Jayan Nagendran, Daniel H Kim, John C Mullen, Holger Buchholz, Philip F Halloran, Gavin Y Oudit
Circulation. 2014;130:A18845
Abstract
Introduction: Heart failure (HF) continues to be a common cause of morbidity and mortality and dilated cardiomyopathy (DCM) is a common cause of adverse ventricular remodeling and HF. Left ventricular assist device (LVAD) has emerged as an important mode of therapy for advanced HF by unloading the LV and maintaining systemic perfusion. The extent to which LVAD mediates beneficial remodeling in the right ventricle (RV) is incompletely understood.
Hypothesis: Differential mRNA expression in a chamber-specific manner may elucidate LVAD mediated functional reverse remodeling in end-stage HF.
Methods and Results: Right (RV) and left (LV) ventricle tissue from explanted failing and non-failing control (NFC) human hearts were collected within 20 mins of explantation. Global mRNA expression profiling in DCM patients without (n=8; 7M/1F; age 42-54 years; ejection fraction (EF): 17.5% (15-26%) and with (n= 8; 7M/1F; age 44-58 yrs; EF 15% (12.5-25.0%) LVAD (HeartMate II) was compared to NFC (n=6; age 39-56 yrs; 4M/2F). mRNA microarray was performed using Affymetrix PrimeView gene chips and evaluated using the Ingenuity Pathway Analysis (IPA) program. LVAD use was associated with marked reduction of myocardial hypertrophy, interstitial fibrosis and apoptosis in both the LV and RV. The cleaved caspase-3 and Tunnel-positive cardiomyocytes were reduced in both RV and LV with LVAD. Total gene entities of 991 in the LV and 860 in the RV were differentially expressed in hearts with DCM without LVAD. LVAD use was independently associated with the altered regulation of 280 genes in the LV and 380 genes in the RV. The total up/down regulated genes involved in cardiac hypertrophy ranged from 20-60% and myocardial fibrosis ranged from 10-80% and were altered in both the RV and LV in response to LVAD therapy. In addition, 14-30% of the total apoptotic-related genes and 6-20 % of the cytokine-related genes were changed in both ventricles.
Conclusions: LVAD reduced pathological hypertrophy and fibrosis in both LV and RV and is correlated with altered gene expression controlling adverse myocardial remodeling and fibrosis, apoptotic and inflammatory pathways. LVAD therapy represents a viable long-term therapy for HF patients and is associated with improved RV remodeling.
Article Information
vol. 130 no. Suppl 2 A18845
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Nirmal Parajuli1;
- Ratnadeep Basu2;
- Konrad S Famulski3;
- Jayan Nagendran4;
- Daniel H Kim2;
- John C Mullen5;
- Holger Buchholz6;
- Philip F Halloran7;
- Gavin Y Oudit1
- 1Medicine, Div of Cardiology, Mazankowski Alberta Heart Institute, Univ of Alberta, Edmonton, Canada
- 2Medicine, Div of Cardiology, Mazankowski Alberta Heart Institute, Univ of Alberta, Edmonton, Canada
- 3Medicine, Div of Nephrology & Transplantation Immunology, Univ of Alberta, Edmonton, Canada
- 4Medicine, Mazankowski Alberta Heart Institute, Div of Cardiac Surgery, Univ of Alberta, Edmonton, Canada
- 5Medicine, Mazankowski Alberta Heart Institute, Div of Cardiac Surgery, Univ of Alberta, Edmonton, Canada
- 6Medicine, Dept of Pediatrics, Stollery Children’s Hosp, Univ of Alberta, Edmonton, Canada
- 7Medicine, Div of Nephrology & Transplantation Immunology, Univ of Alberta, Edmonton, Canada
Abstract 18066: Elevated Levels of Circulating MicroRNA-223, MicroRNA-127 and MicroRNA-146a are Associated with Risk of Near Future Cardiovascular Events in Asymptomatic Individuals
Anna P Pilbrow, Chris M Frampton, A M Richards, Richard W Troughton, Vicky A Cameron
Circulation. 2014;130:A18066
Abstract
Background: MicroRNAs (miRNAs) are small regulatory RNAs that are stable in plasma and may have utility as diagnostic markers in cardiovascular disease.
Aim: This study investigated whether circulating miRNAs predict risk of incident cardiovascular events in the general population, by analyzing associations between plasma levels of 10 miRNAs and fatal/non-fatal cardiovascular events in asymptomatic individuals selected from a community based cohort.
Methods: miRNA levels were measured in 71 individuals, 37 who had a cardiovascular event within 1 year (mean age=70 ±10 years, 65% male) and 34 matched controls without events (68 ±9 years, 59% male). miRNAs were selected from those previously associated with incident myocardial infarction (miR-126 -197 -223), unstable plaque (miR-127 -133a -145) or stable coronary artery disease (miR-142 -146a -323a -652). Associations between miRNAs, NT-proBNP, Framingham risk score and cardiovascular events were tested with Cox regression and log-rank tests.
Results: All miRNAs except miR-323a were positively associated with cardiovascular events, independent of NT-proBNP and Framingham risk score, with miR-223 showing the strongest association (hazard ratio [95% confidence interval] upper vs lower tertile: miR-223 3.7 [1.6-8.7] p=0.002; miR-127 3.7 [1.6-8.5] p=0.003; miR-146a 3.5 [1.4-8.5] p=0.006; miR-142 3.6 [1.5-8.6] p=0.005; miR-197 3.4 [1.4-8.1] p=0.007; miR-145 1.9 [1.2-2.9] p=0.007; miR-652 2.9 [1.2-6.9] p=0.017; miR-133a 2.5 [1.1-5.5] p=0.027; miR-126 2.4 [1.0-5.4] p=0.041). All miRNAs were correlated with ≥1 other miRNA (Pearson correlation >0.6, p<0.001) and no miRNA remained predictive of events once miR-223 was included in the model. A risk score combining miR-223, miR-127 and miR-146a (Figure 1) was a stronger predictor than miR-223 alone.
Conclusions: These data suggest that circulating miR-223, miR-127 and miR-146a may improve prediction of cardiovascular events in asymptomatic individuals.
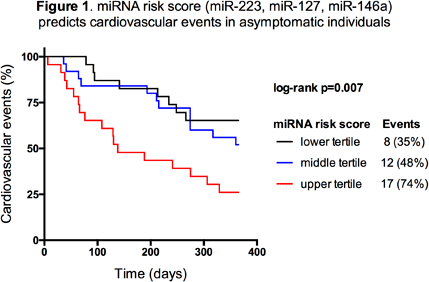
Article Information
vol. 130 no. Suppl 2 A18066
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Anna P Pilbrow;
- Chris M Frampton;
- A M Richards;
- Richard W Troughton;
- Vicky A Cameron
- Medicine, Univ of Otago Christchurch, Christchurch, New Zealand
Abstract 16379: DNA Methylation Profiling Identifies Novel Epigenetic Markers of Cardiovascular Events
Svati H Shah, Lydia C Kwee, Elizabeth A Grass, William E Kraus, Simon G Gregory
Circulation. 2014;130:A16379
Abstract
Objectives: Considerable effort has been made in identifying the genetic underpinnings of cardiovascular disease (CVD), including cardiovascular events. However, DNA variation does not fully account for all heritable factors of CVD risk or early mortality. DNA methylation is a non-DNA sequence-based, heritable form of gene regulation that plays a role in CVD. To identify the role of epigenetic variation in CVD we performed DNA methylation profiling in a CVD cohort (CATHGEN).
Methods: The study population included individuals from the CATHGEN biorepository referred for cardiac catheterization for concern of ischemic heart disease. Cases were defined as individuals who died >580 days (N=62) after index cath. and controls were alive and without any CVD events for >1700 days (N=49) after index cath. We used Illumina’s HumanMethylation450 array to profile the methylation status of 485,000 CpG dinucleotides. After QC, preprocessing, normalization, and removal of cross-reactive probes, we logit-transformed the resulting β-values of the remaining 445,323 probes. We tested for association between methylation and case-control status using a linear model, adjusting for age, race, sex, row and chip. Follow-up was based either on the resulting p-value for association between methylation and case-control status (p>0.001, n=1438 probes), or a threshold for the difference between average methylation status in cases and controls
Article Information
vol. 130 no. Suppl 2 A16379
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Svati H Shah;
- Lydia C Kwee;
- Elizabeth A Grass;
- William E Kraus;
- Simon G Gregory
- Medicine, Duke Molecular Physiology Institute, Durham, NC
Abstract 15634: Epigenetic Modifiers Reduce Inflammation and Modulate Macrophage Phenotype to Attenuate Endotoxemia-Induced Acute Lung Injury
Bahar Barani, Jayakumar Thangavel, Sheeja Rajasingh, Yu-Ting Xuan, Buddhadeb Dawn, Johnson Rajasingh
Circulation. 2014;130:A15634
Abstract
Introduction: Acute lung injury (ALI) during sepsis is characterized by bilateral alveolar infiltrates, lung edema, and respiratory failure. Although chemical epigenetic modifiers can potentially limit lung inflammation, to date no study has examined the efficacy of DNA methyl transferase (DNMT) inhibitor Aza (5-Aza 2-deoxycytidine) and histone deacetylase (HDAC) inhibitor TSA (Trichostatin A) combination therapy (Aza+TSA) on inflammation, apoptosis and survival in sepsis.
Hypothesis: Therapy with Aza+TSA will modulate cytokines and macrophages to quench inflammation in ALI.
Methods and Results: In an LPS-induced ALI mouse model, a single dose of Aza+TSA prevented lung inflammation and injury with significant reduction in mortality as compared with mice treated with either Aza or TSA alone. A significant attenuation of adverse lung histopathological changes, and inflammation was noted. RT-PCR array data showed that LPS-induced primary macrophages expressed mRNA transcripts of pro-inflammatory chemokines (ccl2, ccl3, ccl4, ccl5, ccl7, CD40, cxcl10 and cxcl12) and cytokines (IL-1α, IL-1β, IL-1R, IL-6, IL-18, ITGβ2, lymphotoxin-β and TNFα). These proinflammatory molecules were significantly reduced after treatment with Aza+TSA. We also observed significantly higher levels of IL-10 and IL-10R mRNA expression in LPS-induced macrophages treated with Aza+TSA rather than either drug alone. Importantly, our FACS data showed a significant increase in the expression of M2 markers CD23 and CD124 in LPS-induced macrophages treated with Aza+TSA compared with untreated LPS-stimulated cells and control cells (Figure).
Conclusions: Combinatorial treatment with Aza+TSA reduces inflammation and promotes an anti-inflammatory M2 macrophage phenotype in the lung during sepsis. This novel epigenetic therapy has therapeutic potential for patients with sepsis and ALI.
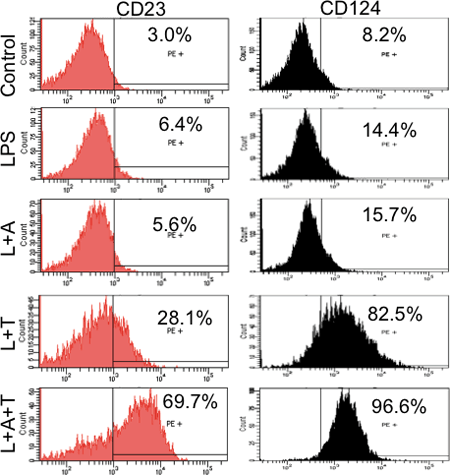
Article Information
vol. 130 no. Suppl 2 A15634
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Bahar Barani;
- Jayakumar Thangavel;
- Sheeja Rajasingh;
- Yu-Ting Xuan;
- Buddhadeb Dawn;
- Johnson Rajasingh
- Cardiovascular Diseases, Cardiovascular Rsch Institute, Univ of Kansas Med Cntr, Kansas City, KS
Abstract 14050: The Gut Flora Promotes Endothelial Dysfunction Through Vascular MicroRNA-204-mediated Down-regulation of Endothelial Sirtuin1
Ajit Vikram, Young-Rae Kim, Santosh Kumar, Julia S Jacobs, Kaikobad Irani
Circulation. 2014;130:A14050
Abstract
The gut flora contributes to development of atherosclerosis. Endothelial dysfunction, one manifestation of which is impaired endothelium-dependent vasorelaxation, accompanies and promotes atherosclerotic vascular disease. Here we show that gut flora impair endothelium-dependent vasorelaxation by remotely up-regulating microRNA-204 (miR-204) which downregulates SIRTUIN1 (SIRT1) in the vascular wall. Microarray analysis in aortas of germ-free mice revealed a set of down-regulated microRNAs, including miR-204, which target SIRT1. Suppression of gut flora in mice with antibiotics in drinking water decreased aortic miR-204, increased aortic SIRT1, and improved endothelium-dependent vasorelaxation, effects that were reversed with discontinuation of antibiotics. In addition, miR-204 mimic impaired endothelium-dependent aortic vasorelaxation ex vivo. Moreover, high-fat diet feeding stimulated aortic miR-204, suppressed SIRT1, and impaired endothelial function, all of which were mitigated by administration of antibiotics, and reversed with stoppage of antibiotics. In contrast, antibiotics did not improve high-fat diet-induced endothelial dysfunction in mice conditionally lacking endothelial SIRT1. In addition, anti-miR-204 delivered systemically prevented high-fat diet-induced endothelial dysfunction and vascular SIRT1 decrease. Finally, serum from mice on antibiotics suppressed miR-204, and increased SIRT1, in endothelial cells, effects that were not observed with serum from mice in which antibiotics were discontinued. Therefore, the gut flora remotely downregulates endothelial SIRT1 through miR-204, leading to impairment of endothelial function.
Article Information
vol. 130 no. Suppl 2 A14050
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Ajit Vikram;
- Young-Rae Kim;
- Santosh Kumar;
- Julia S Jacobs;
- Kaikobad Irani
- Internal Medicine, Univ of Iowa, Iowa City, IA
Abstract 13338: A Molecular Signature with the Potential for Identifying Impending Acute Myocardial Infarction
Evan D Muse, Haiying Wang, Paddy Barrett, Fereshteh Parviz, Mark A Novotny, Roger S Lasken, Timothy A Jatkoe, Mark C Connelly, Kurt M Schilling, Chandra Rao, Ali Torkamani, Eric J Topol
Circulation. 2014;130:A13338
Abstract
The inability to temporally and accurately predict acute myocardial infarction (AMI) impairs our ability to further improve patient outcomes. Prior work investigating the potential of circulating endothelial cells (CECs) as a biomarker for AMI has focused mainly on increased enumeration in AMI. Here we describe the designation of a specific gene expression pattern acting as a molecular signature for AMI present in whole blood of patients that was determined using microarray analysis of enriched CECs. Using CELLSEARCH® technology we first separated and enriched CECs from the peripheral blood of patients with AMI (n=53) or from healthy controls (n=53). Total RNA was analyzed using the Affymetrix Human U133 Plus 2.0 Microarray for the expression of over 47,000 transcripts. The expression profiles were examined to determine which genes were selectively up-regulated in AMI compared to controls. We nominated 19 candidate genes from the microarray analysis that demonstrated the ability to discriminate between AMI and controls. To determine if these discriminatory profiles can be detected in samples processed without CEC enrichment, we performed qRT-PCR on the candidate genes using the whole blood of patients with AMI (n=46) and healthy controls (n=30). Heparin-binding EGF-like growth factor (HBEGF) showed the highest discriminatory performance between MI and controls (AUC 0.961, 0.887-0.992, p< 0.0001) in the whole blood analysis. This was followed by NR4A2 (AUC 0.886), NR4A3 (AUC 0.863), EFEMP1 (AUC 0.888), NFKBIA (AUC 0.814) and NLRP3 (AUC 0.811) (p<0.0001 for all). A linear combination of the weighted scores for these 6 genes discriminated AMI from controls with an AUC of 0.97 (p<0.0001). Further study will determine if this proposed six gene molecular signature will lead to the first gene-based, point-of-care testing for the prediction of AMI in an at-risk population.
Article Information
vol. 130 no. Suppl 2 A13338
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Evan D Muse1;
- Haiying Wang2;
- Paddy Barrett1;
- Fereshteh Parviz3;
- Mark A Novotny4;
- Roger S Lasken4;
- Timothy A Jatkoe5;
- Mark C Connelly3;
- Kurt M Schilling6;
- Chandra Rao3;
- Ali Torkamani1;
- Eric J Topol1
- 1Scripps Translational Science Institute, The Scripps Rsch Institute, La Jolla, CA
- 2Rsch and Development, Ortho Clinical Diagnostics, Raritan, NJ
- 3Oncology Biomarkers, Janssen Rsch & Development, LLC, Huntingdon Valley, PA
- 4Single Cell Genomics, J. Craig Venter Institute, La Jolla, CA
- 5Global Med and Scientific Affairs, Janssen Diagnostics, LLC, Raritan, NJ
- 6Rsch and Development, Ortho Clinical Diagnostics, Rochester, NY
Abstract 11433: Postinfarction Cardiac Remodeling Normally Proceeds in Granulocyte Colony-Stimulating Factor Knockout Mice
Kentaro Morishita, Genzou Takemura, Akiko Tsujimoto, Hiromitsu Kanamori, Itta Kawamura, Atsushi Mikami, Hideshi Okada, Toshiaki Takeyama, Tomonori Kawaguchi, Takatomo Watanabe, Kazuko Goto, Atsushi Ogino, Megumi Morishita, Hiroaki Ushikoshi, Masanori Kawasaki, Shinji Ogura, Shinya Minatoguchi, Shinya Minatoguchi
Circulation. 2014;130:A11433
Abstract
Treatment with granulocyte colony-stimulating factor (G-CSF) has been reported to mitigate postinfarction cardiac remodeling. We here examined how the postinfarction remodeling proceeds in the hearts of G-CSF-null mice, of which outcome we hypothesized to be miserable. We generated myocardial infarction in G-CSF-knockout (KO) mice and wild-type (WT) mice by ligation of the left coronary artery. The acute infarct size was similar at 24 h after ligation between the groups, where the incidence of in situ nick-end labeling (TUNEL)-positive cardiomyocytes was similar while the infiltrating leukocytes were significantly fewer in KO mice (n = 6 each). Somewhat surprisingly, at the chronic stage (4 weeks postinfarction), there was no difference in the left ventricular dimension, function and histological findings including the infarct size and myocardial vascular density between the groups (n = 6 each). Because it was contrary to our hypothesis, we next investigated the changes in major angiogenesis growth factors such as angiopoientin-1, hepatocyte growth factor, hypoxia induced factor-1 alpha, insulin-like growth factor-1, vascular endothelial growth factor (VEGF) in postinfarction hearts. As a result, biochemical analyses revealed a markedly increased expression of VEGF in the hearts of KO mice compared with WT mice (n = 6 each). The findings suggest a possibility that overexpressed VEGF might have compensated for the defect of G-CSF in KO mice. To check this hypothesis, we next inhibited the VEGF signal using an anti-VEGF antibody bevacizumab. Then the cardiac remodeling was significantly aggravated at the chronic stage, and the immunostaining showed scarce vascular development, reduced cell proliferation activity, and increased apoptosis in the granulation tissue of the bevacizumab-treated hearts at the subacute stage (4 days postinfarction), which might have contributed to the aggravation of cardiac remodeling during the chronic stage. In conclusion, G-CSF-KO mice unexpectedly displayed normal process of postinfarction cardiac remodeling. Overexpressed VEGF in the KO mice is suggested to compensate for the deficit of G-CSF through enhancement of neovascularization in the postinfarction heart.
Article Information
vol. 130 no. Suppl 2 A11433
Published By:
American Heart Association, Inc.
Online ISSN:
History:
- Originally published November 14, 2014.
Copyright & Usage:
© 2014 by American Heart Association, Inc.
Author Information
- Kentaro Morishita1;
- Genzou Takemura2;
- Akiko Tsujimoto1;
- Hiromitsu Kanamori1;
- Itta Kawamura1;
- Atsushi Mikami1;
- Hideshi Okada1;
- Toshiaki Takeyama1;
- Tomonori Kawaguchi1;
- Takatomo Watanabe1;
- Kazuko Goto1;
- Atsushi Ogino1;
- Megumi Morishita1;
- Hiroaki Ushikoshi1;
- Masanori Kawasaki1;
- Shinji Ogura3;
- Shinya Minatoguchi1;
- Shinya Minatoguchi1
- 1Dept of Cardiology, Gifu Univ Graduate Sch of Medicine, Gifu, Japan
- 2Div of Internal Medicine, Asahi Univ Sch of Dentistry, Gifu, Japan
- 3Emergency and Disaster Medicine, Gifu Univ Graduate Sch of Medicine, Gifu, Japan